CRISPR-Cas: Un Nobel y una esperanza para tratar las enfermedades genéticas
Introducción
El ser humano existe y funciona gracias al encendido [1] ordenado y coordinado de miles de genes. Éstos son secuencias de ADN que contienen la información necesaria para generar una característica física (rasgo) o bioquímica (proteína). Los cambios en las secuencias de ADN (mutaciones) de nuestros genes nos dan diversidad; también pueden ser la causa de que algo (ARN o proteínas) deje de funcionar correctamente. Las enfermedades causadas por mutaciones en los genes de los pacientes se conocen como enfermedades genéticas. A continuación, aprenderás cómo la mutación de un gen específico hace que una súper proteína, llamada hemoglobina, se vuelva defectuosa. Esta hemoglobina defectuosa es el origen de una enfermedad llamada anemia falciforme. Las personas con este padecimiento tienen baja expectativa de vida. Gracias al descubrimiento de un tipo de tijeras moleculares en las bacterias, el ADN de las células puede cortarse y manipularse para curar a los pacientes con anemia falciforme, entre otras enfermedades.
La anemia falciforme y la regulación de genes
La anemia falciforme es una enfermedad crónica causada por una mutación en el gen HBB, el cual contiene la información para producir la proteína β-globina (beta) adulta. Dos proteínas β-globina se asocian con dos proteínas α-globina (alfa) para producir una súper proteína globular llamada hemoglobina adulta (hemoglobina A, HbA). Imaginemos a las proteínas globinas como piezas de rompecabezas. Dos piezas idénticas, moradas, se juntan con dos partes iguales, rojas, para formar la hemoglobina (Fig. 1). Ésta se encuentra en los eritrocitos (glóbulos rojos) de la sangre; cuando pasan por los pulmones, la hemoglobina atrapa el oxígeno disponible y lo transporta a todas las células del cuerpo través del torrente sanguíneo. Así, una hemoglobina sana hace funcionar correctamente a tejidos, órganos y sistemas.
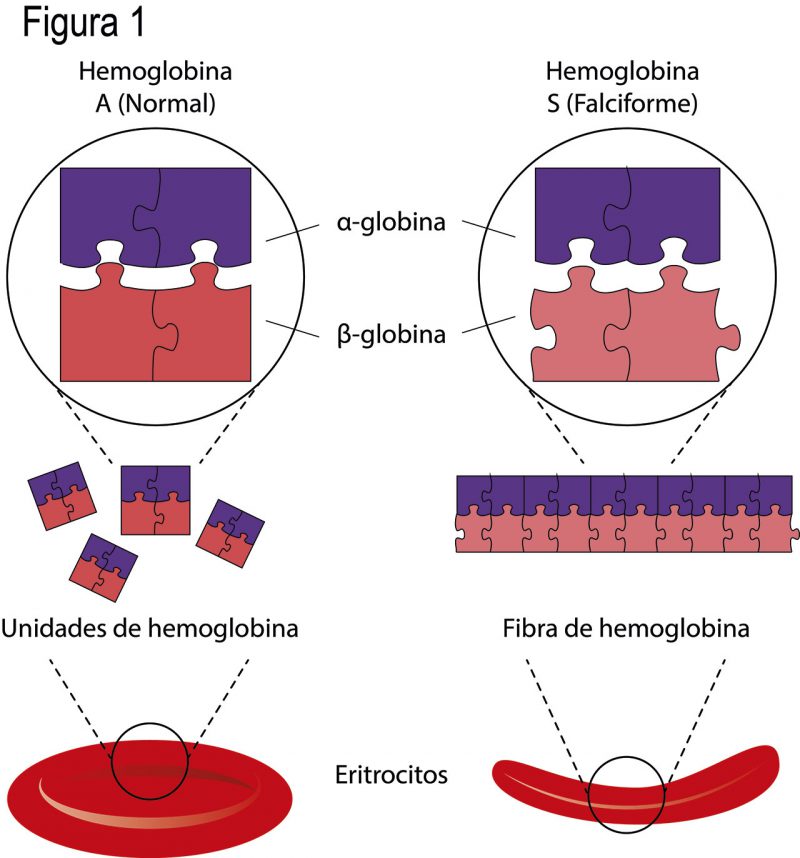
La mutación en HBB de los pacientes con anemia falciforme produce una β-globina con una estructura anormal (Fig. 1, rosa) que se asocia de manera diferente a las α-globinas, provocando que se forme una fibra larga (cadena) de hemoglobinas (HbS) cuando liberan el oxígeno a las células (Fig. 1). Las fibras de hemoglobina tienen menor capacidad de captar oxígeno, además, afectan la forma y fisiología de los eritrocitos. Cuando éstos son normales tienen una forma similar a la de una dona y se deforman fácilmente; mientras que los anémicos adquieren un aspecto como de hoz y son muy rígidos (Fig. 1). Esta rigidez llega a producir aglomeraciones de eritrocitos en los pequeños capilares sanguíneos, obstruyendo el flujo de la sangre y causando menor disponibilidad de oxígeno, inflamación, dolor y hasta microinfartos.
Los tratamientos más eficaces para los pacientes de anemia falciforme, son transfusiones sanguíneas frecuentes (que contienen eritrocitos sanos), o los trasplantes de médula ósea (cuyas células troncales hematopoyéticas dan origen a los eritrocitos). Las transfusiones sanguíneas ayudan, principalmente, en casos agudos. Por otro lado, el trasplante de médula ósea es -o era- el único remedio para la anemia falciforme; sin embargo, es muy difícil conseguir donadores cuyos antígenos leucocitarios sean compatibles con los del paciente.
La anemia falciforme se identifica entre los 4 y 6 meses de vida de un ser humano. ¿Por qué una enfermedad con síntomas tan severos se detecta meses después del nacimiento? Ello se debe a un fenómeno fisiológico conocido como recambio de la hemoglobina. Todos los vertebrados presentamos distintos tipos de hemoglobina conforme transcurre el desarrollo embrionario. En nuestra especie, las globinas ζ (dseta) y ε (épsilon) constituyen la hemoglobina embrionaria, la cual se encuentra en las primeras 6 semanas de gestación. Posteriormente, las globinas α y γ (gama) forman la hemoglobina fetal (HbF), que es la principal responsable del transporte de oxígeno en el feto, desde la semana 6 de gestación hasta 4 semanas después del nacimiento (Fig. 2). Después de las 4 semanas de nacimiento, la hemoglobina A, formada por las globinas α y β, va reemplazando a la hemoglobina fetal hasta volverse la molécula principal de transporte de oxígeno (Fig. 2). Sin embargo, nuestro cuerpo contiene cantidades considerables de hemoglobinas fetales (> 20% del total de globinas) hasta los 6 meses del nacimiento.
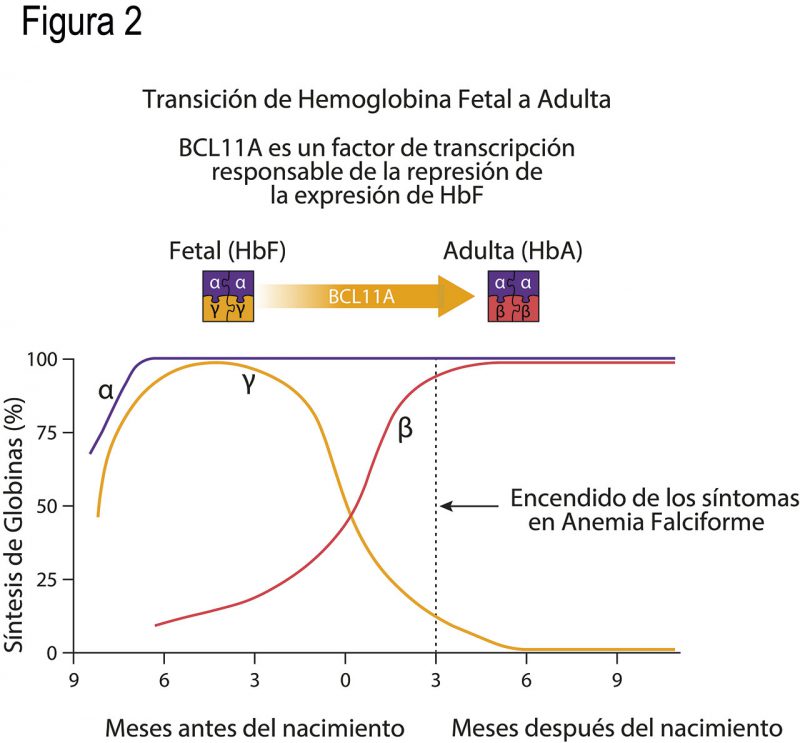
El recambio de la hemoglobina sucede a través de un mecanismo altamente controlado, de “encendido y apagado” gradual de los genes que codifican para las distintas proteínas globinas. La razón por la que los bebés con anemia falciforme no presentan síntomas hasta después de 4 – 6 meses de nacidos es que el gen de la γ-globina no se ha apagado por completo (Fig. 2). Así, las cantidades de γ-globina presentes en este periodo postnatal son suficientes para frenar la formación de fibras largas de hemoglobina y, con ello, tener eritrocitos funcionales. Si gracias a la γ-globina los bebés tienen una vida normal en sus primeros meses, ¿por qué no simplemente sigue prendido este gen el resto de la vida de ese ser humano? [2]
Los mecanismos moleculares que intervienen en el silenciamiento del gen HBG de la γ-globina son distintos. En 2008, un grupo de investigación en la Universidad de Harvard identificó a la proteína BCL11A como actor principal en el apagado del gen HBG. En ese momento, varios laboratorios se dedicaron a estudiar al gen BCL11A. Descubrieron un potenciador [3] que regula la actividad de ese gen. Al mutar este potenciador, o eliminarlo, se disminuye la cantidad de proteína BCL11A en las células estudiadas. Asimismo, los niveles de la γ-globina se incrementaron en dichas células mutantes. Al repetir esos experimentos en ratones modelo de anemia falciforme, comprobaron que sus síntomas disminuían considerablemente y su comportamiento se asemejaba al de ratones sanos. Para reducir las afecciones de la anemia falciforme basta con mantener la γ-globina disponible, pero para ello hay que reducir los niveles de BCL11A.
Busca en las bacterias
Diariamente se producen descubrimientos motivados por la curiosidad de conocer cómo funciona una minúscula parte del todo que es el universo. Lo más extraordinario ocurre cuando esos pequeños hallazgos sientan las bases del desarrollo de tecnologías que no fueron imaginadas al inicio de una investigación. Fue el caso de la revelación de la transformación bacteriana y la síntesis de proteínas, que permite producir insulina en bacterias; de la invención de la PCR (reacción en cadena de la polimerasa), empleada ampliamente en criminalística, pruebas de paternidad, y, recientemente, en la detección del SARS-CoV2 en muestras de pacientes con síntomas de COVID. También fue el caso del desarrollo del sistema de edición de genes CRISPR-Cas, que se explica a continuación.
A fines de los años 90 y principios del 2000, Francisco Mojica, un notable científico de la región de Alicante, España, descubrió una región en el genoma de distintos microorganismos, la cual contenía varias series de secuencias cortas de ADN repetidas [4] y palindrómicas [5]. Una de las características particulares de la región es que los fragmentos de repetidas siempre se encontraban espaciados por secuencias únicas de alrededor de 30 pares de bases (Fig. 3). Lo que aumentó la curiosidad por estas regiones fue el hallazgo de que los fragmentos de secuencias espaciadoras eran idénticos (homólogos) a los encontrados en los genomas de bacteriófagos (virus que infectan bacterias y arqueas). Además, el Dr. Mojica se percató de que si una especie de bacteriófago X tenía fragmentos de su genoma embebidos en la región de repetidas conservadas de una cepa bacteriana Y; X ya no podía infectar a Y (Fig. 3). Esto llevó a Mojica a proponer que esa región conservada de secuencias repetidas funcionaba como un sistema inmune adaptativo en bacterias y arqueas.
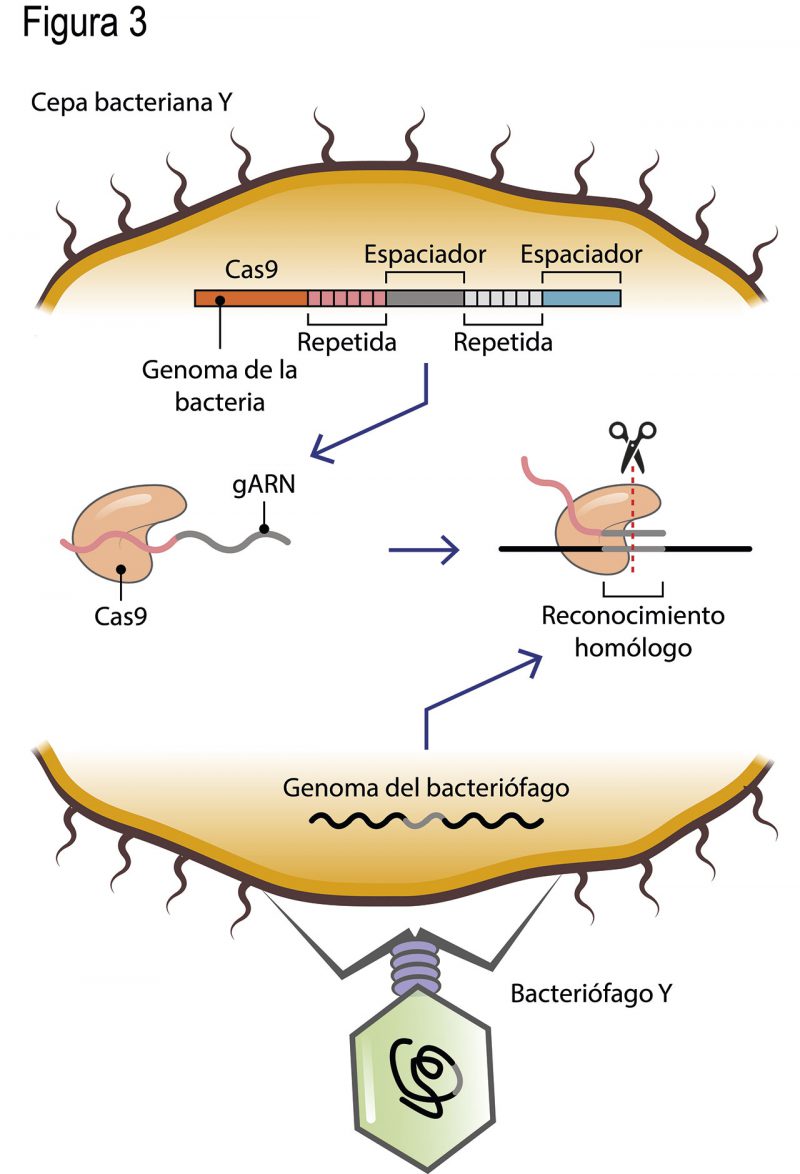
Francisco Mojica nombró a esta región, tan particular y conservada en tantos microorganismos, como “CRISPR” por sus siglas en inglés Clustered Regularly Interspaced Short Palindromic Repeats, que significan Repetidas Palindrómicas Cortas Agrupadas y Regularmente Interespaciadas. La propuesta de un sistema inmune adaptativo en los microorganismos llevó a una decena de grupos de investigación en el mundo a intentar descifrar su funcionamiento [6]. Descubrieron que las secuencias provenientes de virus, y las repetidas adyacentes, se transcriben en una molécula larga de ARN que es procesada en fragmentos de ARN más pequeños, que contienen la secuencia de virus más la secuencia repetida. Las secuencias de virus se denominan espaciadores y, al ser idénticas a fragmentos del genoma del virus, se aparean con su secuencia homóloga; es decir, sirven para reconocer al invasor (Fig. 3). La secuencia repetida funciona como una acarreadora de un tipo de tijeras moleculares (la endonucleasa Cas9) que cortan el genoma del virus sólo dónde el espaciador lo indica (Fig. 3).
Una de las características del sistema CRISPR-Cas que llamó la atención de los investigadores fue que el corte del genoma del fago invasor se produce sólo en la región homóloga (protoespaciador) al espaciador. Es decir, una pequeña secuencia de 20 – 30 nucleótidos de ARN guía le indica a la proteína Cas9 dónde cortar a una molécula de ADN. ¿Se podrían sintetizar secuencias cortas de ARN para cortar cualquier región genómica, de cualquier organismo, con las tijeras Cas9? Esta pregunta se la hicieron las Dras. Emmanuelle Charpentier y Jennifer Doudna, a partir del conocimiento que tenían del sistema inmune adaptativo en bacterias y arqueas.
Las tijeras moleculares funcionan fuera de las bacterias
Gracias a los avances en biología molecular de los últimos 40 años, pueden sintetizarse moléculas como el ARN en tubos de plástico, o producir y purificar miligramos de proteína como la insulina, a partir de cultivos de la bacteria Escherichia coli. Eso fue lo que hicieron los grupos de investigación de Charpentier y Doudna. Diseñaron y sintetizaron una molécula de ARN parecida a la que guía a la Cas9 en bacterias, con la diferencia principal de que la secuencia correspondiente al espaciador no era similar a la de ningún genoma viral. En cambio, era idéntica a una secuencia de 20 nucleótidos del gen de la proteína verde fluorescente (GFP por sus siglas en inglés) de Aequorea victoria (Fig. 4). Esa molécula de ARN se encargaría de guiar la Cas9 a su secuencia homóloga en el gen de GFP, para realizar un corte, por ello la denominaron guía de ARN (gARN) (Fig. 4).
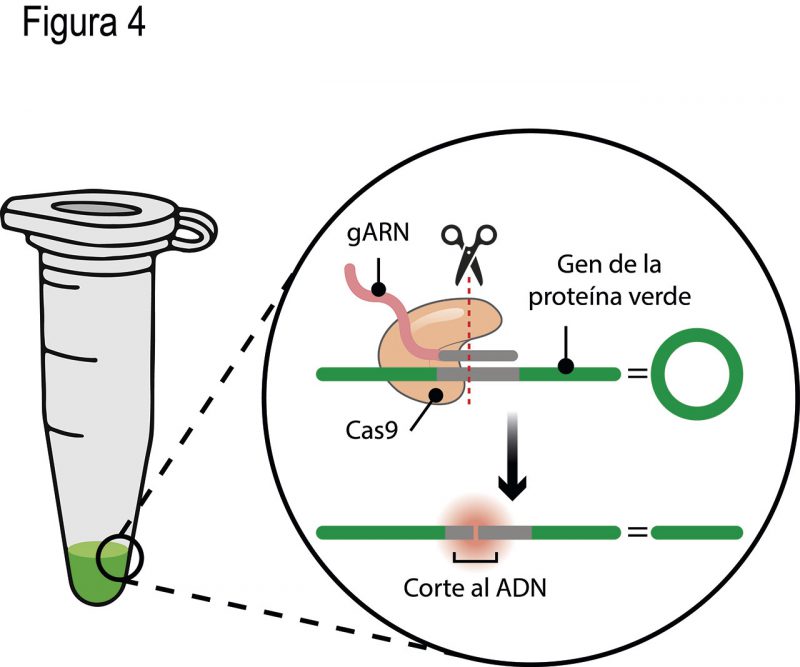
Las científicas, y sus estudiantes, decidieron incubar: el gARN, la proteína Cas9 y un plásmido (cromosoma circular) donador del gen de la GFP, todos juntos en un pequeño tubo de plástico de 1.5 mL (Fig. 4). El resultado fue un plásmido lineal, cortado exactamente en la secuencia homóloga del gARN en el gen de GFP (Fig. 4). Lo que acababan de desarrollar, gracias a los elementos del sistema inmune adaptativo encontrado en bacterias y arqueas, fue una técnica sencilla y versátil de edición genética. Su hallazgo lo publicaron en junio del 2012 en la revista Science.
Lo revolucionario del sistema fue comprobar que unas tijeras moleculares (Cas9) bacterianas, guiadas por pequeñas moléculas de ARN (gARN) hacia secuencias específicas, pueden funcionar fuera de las bacterias para reconocer y cortar casi cualquier secuencia de ADN (Fig. 4). Incluso secuencias de ADN del genoma de hongos, plantas y animales. En octubre de 2020, las Dras. Emmanuel Charpentier y Jennifer Doudna recibieron el Premio Nobel de Química “por el desarrollo de un método para la edición del genoma”.
Editar el genoma se refiere a cambiar la secuencia de nucleótidos en una región específica del ADN; es decir, inducir o introducir mutaciones. El concepto clave es región específica. Distintos agentes externos (rayos X, luz UV, moléculas entrecruzantes, etcétera) pueden generar mutaciones en el genoma, pero ocurren en regiones al azar. En la edición genética, las mutaciones suceden en las secuencias de ADN que los investigadores deciden dentro de las células de un organismo. Los cortes en el ADN son interpretados como daño al ADN. La célula, al percibir que una de sus moléculas de ADN ha sufrido un daño, activa sus mecanismos de reparación del ADN. Durante el proceso, la región de ADN reparada puede perder o ganar nucleótidos, por lo que la secuencia ya no es idéntica, ha mutado.
Para editar el genoma de una célula, basta con meter las tijeras moleculares Cas9 y un gARN, con una secuencia homóloga a la región que se quiere editar, al interior del núcleo de la célula. En febrero de 2013, un conjunto de investigadores, liderados por el Dr. Feng Zhang, comprobaron que el sistema CRISPR-Cas9 funcionaba para editar el genoma de células humanas. A partir de ese momento, se han editado, con CRISPR-Cas, genomas de células de cientos de organismos distintos.
Las investigaciones de Mojica, Charpentier, Doudna, Zhang y un cúmulo de investigadores, hicieron posible identificar una secuencia específica en el genoma de cualquier célula y cortar ahí para producir mutaciones o corregirlas. La ingeniería genética vive, en palabras del famoso científico Eric Lander, “una era revolucionaria en la que las posibilidades están limitadas sólo por nuestra imaginación colectiva”. La plasticidad para generar gARNs y la especificidad de corte que otorga el sistema CRISPR-Cas de edición genética, son las ventajas más poderosas para utilizarlo en un sinfín de aplicaciones. Hay una en especial, que por décadas ha esperado el desarrollo de una tecnología con dichas características: la terapia génica.
Corte y confección para curar la anemia falciforme
La terapia génica es un campo de la medicina con distintos enfoques para tratar pacientes con enfermedades causadas por mutaciones en genes específicos, como la anemia falciforme. Gracias a los descubrimientos sobre BCL11A y su regulación sobre la γ-globina, se sabía que inhibiendo dicho factor se podía obtener una mejor prognosis para los pacientes. Así que, distintos grupos de investigación decidieron emplear las tijeras CRISPR para cortar el potenciador de BCL11A para reducir los niveles del represor en las células troncales pluripotentes que dan origen a los eritrocitos: las células troncales hematopoyéticas CD34+, localizadas en la médula ósea.
Inyectar los ingredientes del sistema CRISPR en la médula ósea sería muy riesgoso y complicado. Los investigadores optaron por realizar una edición de las células CD34+ cultivadas en una caja de Petri, lo cual es una manera mucho más controlada de abordar el problema. Para lograrlo, extrajeron células CD34+ de donantes sanos y, en ellas, realizaron la edición con CRISPR-Cas del potenciador de BCL11A (Fig. 5). Después de comprobar que la edición había ocurrido de manera eficaz y que no se había cortado ninguna otra región del ADN, transfundieron las células troncales pluripotentes CD34+ editadas en un paciente con anemia falciforme (Fig. 5).
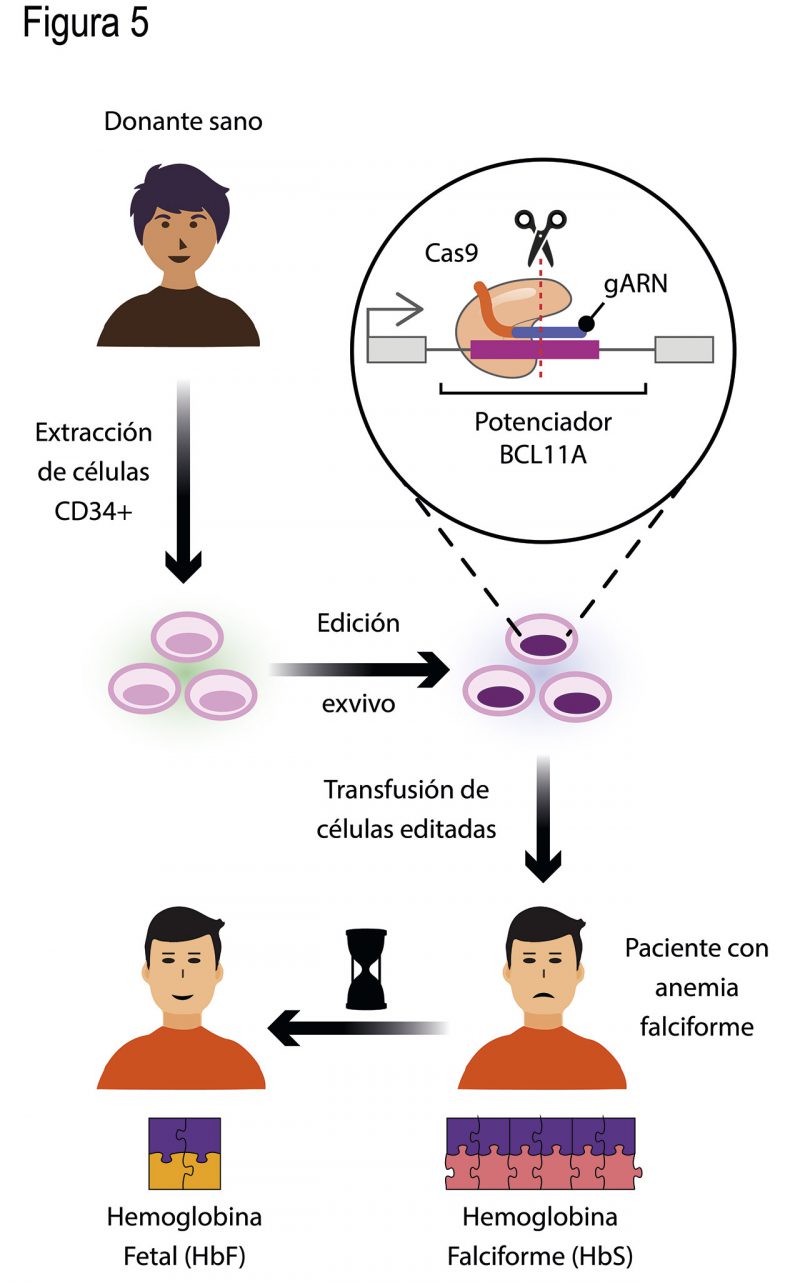
¿Cómo editaron al potenciador BCL11A en las células CD34+? Los investigadores utilizaron un solo gARN con homología de secuencia a una región muy importante para el funcionamiento del potenciador (Fig. 5). Insertaron el coctel de CRISPR en dichas células y evaluaron el resultado. El potenciador de las células CD34+ fue editado, inactivando su función. Han pasado más de dos años de que el paciente con anemia falciforme recibió la transfusión de las células CD34+ editadas. Más del 98% de sus células sanguíneas contienen ahora niveles altos de la hemoglobina fetal (Fig. 5). Esta persona ya no ha necesitado de transfusiones de médula ni ha sufrido por obstrucciones en los vasos sanguíneos.
Aunque el ejemplo anterior podría parecer un caso aislado, representa una prueba de concepto de que los tratamientos, utilizando células editadas con CRISPR-Cas mejoran la salud de pacientes con enfermedades genéticas. Este tratamiento ya se utiliza en pacientes con aterioesclerosis, fenilcetonuria, distrofia muscular de Duchenne, amaurosis congénita de Leber, diabetes, fibrosis quística e, incluso, en enfermedades infeccionas como SIDA. La cura a muchas enfermedades crónicas se encontró en las bacterias.
Notas al pie
[1] Los genes pueden estar en dos estados de acuerdo con su actividad transcripcional (síntesis de ARN). Cuando un gen se transcribe se dice que se encuentra “encendido”; cuando un gen no se transcribe se considera “apagado”.
[2] Existen pacientes con niveles constantes y relativamente altos de hemoglobina fetal. Normalmente tienen síntomas menos severos relacionados con su anemia falciforme. Muchos de ellos tienen mutaciones en el potenciador3 de BCL11A, que inactivan la función. Por lo tanto, los niveles de expresión del represor BCL11A son muy bajos, permitiendo que la γ-globina se exprese durante toda la vida del paciente.
[3] Un potenciador es una secuencia de ADN que “ayuda” a incrementar la transcripción de un gen, o genes, específico(s).
[4] Se dice que una secuencia de ADN es repetida cuando una serie específica de nucleótidos ordenados se encuentra varias veces en el genoma de un organismo; por ejemplo, en la secuencia AGTAGCCGATTATGTAGCCGATTC, el fragmento GTAGCCGATT la podemos encontrar subrayada dos veces.
[5] Se dice que una secuencia de ADN es palíndrome cuando se lee de igual forma al derecho que al revés.
[6] Las bacterias y arqueas han competido con los batecteriófagos por millones de años, desarrollando distintas estrategias de defensa a las infecciones. Lo extraordinario del mecanismo CRISPR-Cas es que es adaptativo y específico. Las bacterias, que sobreviven a una infección por una especie de fago, integran fragmentos de secuencias (20 – 30 nt) provenientes del genoma invasor en su región de repetidas CRISPR. De esa manera pueden “recordar” a esa especie de virus en una futura invasión.
Referencias
Barbarina G., Labedz A., Stucchi S., Abbiati A. & Ronchi A. E. (2021) Physiological and Aberrant γ-Globin Transcription During Development. Front Cell Dev Biol; 9:640060. DOI: 10.3389/fcell.2021.640060.
Frangoul H., Altshuler D., Cappellini M. D., Chen Y. -S., Domm J., Eustace B. K., Foell J., de la Fuente J., Grupp S., Handgretinger R., Ho T. W., Kattamis A., Kernystsky A., Lekstrom-Himes J., Li A. M., Locatelli F., Mapara M. Y., de Montalembert M., Rondelli D., Sharma A., Sheth S., Soni S., Steinberg M. H., Wall D., Yen A. & Corbacioglu S. (2021) CRISPR-Cas9 Gene Editing for Sickle Cell Disease and β-Thalassemia. N Engl J Med; 384:252-60. DOI: 10.1056/NEJMoa2031054.
Sankaran V. G., Menne T. F., Xu J., Akie T. E., Lettre G., Van Handel B., Mikkola H. K. A., Hirschhorn J. N., Cantor A. B. & Orkin S. H. (2008) Human Fetal Hemoglobin Expression is Regulated by the Developmental Stage-Specific Repressor BCL11A. Science; 322:1839-1842. DOI: 10.1126/science.1165409.