Estudio de la estructura secundaria de proteínas y sus modificaciones
La luz interactúa continuamente con la materia y la curiosidad por el estudio de este tipo de eventos se remonta a tiempos lejanos. En particular, los fenómenos quirópticos llamaron la atención de científicos notables como Jean-Baptiste Biot y Augustin Fresnel hace poco más de dos siglos. Estos fenómenos tienen su origen en la quiralidad de ciertas moléculas que comparten composiciones químicas idénticas y presentan estructuras tridimensionales (3D) que son la imagen especular una de la otra. Es decir, guardan una relación de simetría como la que existe entre las manos izquierda y derecha. Ya en 1848, Louis Pasteur demostró el fenómeno conocido como actividad óptica mediante un experimento sencillo e ingenioso; con paciencia meticulosa y empleando únicamente identificación visual, Pasteur logró separar pequeños cristales formados por isómeros de tartrato de sodio y amonio, de acuerdo con su quiralidad. Al disolverlos por separado en agua, cada solución hizo rotar de forma opuesta a un haz de luz polarizada.
Durante buena parte del siglo pasado y lo que va de éste, se han estudiado ampliamente los fenómenos de absorción y emisión de la radiación electromagnética (que puede ser luz visible) debido a su interacción con la materia, lo que ha dado lugar a diferentes tipos de espectroscopía. Esta interacción depende fuertemente, tanto de la estructura interna del material en estudio, como de la longitud de onda (o energía) de la radiación incidente. Nos interesa de forma especial la espectroscopía conocida como dicroísmo circular (DC), que consiste en el estudio de la absorción diferencial de dos ondas electromagnéticas con polarización circular izquierda y derecha. Si las moléculas estudiadas cuentan con centros quirales, entonces se obtiene un espectro de DC.
El estudio de algunas de las biomoléculas más importantes, como los carbohidratos, las proteínas o los ácidos nucleicos, es de gran interés ya que poseen átomos de carbono quirales. En el caso de las proteínas, los residuos en la cadena polipeptídica se pueden organizar en diferentes motivos estructurales, originados por las diferentes posibilidades de formación de puentes de hidrógeno entre los átomos del esqueleto de la proteína. A este nivel de ordenamiento se le conoce como estructura secundaria. Entre las estructuras más conocidas se encuentran las hélices alfa, las hojas betas, los giros y las conformaciones desordenadas. Sin embargo, debe mencionarse que existen algunas otras variantes estructurales. Una propiedad interesante de las estructuras secundarias es su quiralidad intrínseca (por ejemplo, la mayor parte de las hélices alfa son dextrógiras y una minoría son levógiras), y cada una de estas estructuras posee un espectro característico de DC, cuyo intervalo de longitudes de onda corre típicamente entre 180 y 250 nm. Es decir, dicho espectro se encuentra localizado en la región del ultravioleta lejano. De esta forma, la espectroscopía DC permite identificar y asignar la estructura secundaria.
Cabe mencionar que los experimentos de DC requieren de una pequeña cantidad de muestra y además se llevan a cabo con rapidez, lo que permite estudiar cambios estructurales en términos de la temperatura, de variaciones del pH del medio o de la concentración de cosolutos. También es importante conocer si hay cambios estructurales por reemplazo de residuos o bien estudiar la similitud de nuevas proteínas con proteínas análogas ya conocidas. Todas estas propiedades resultan más relevantes cuando es posible comparar el espectro experimental con un espectro de DC calculado a partir de la estructura atómica 3D de la proteína en cuestión. Esta última puede obtenerse a partir de una base de datos como Protein Data Bank (PDB) [1], de una simulación numérica o de algoritmos de predicción estructural como AlphaFold 3 (AF3) [2], I-Tasser (I-Ta) [3] o Rosetta (Ros) [4]. Cabe destacar que el Premio Nobel de Química 2024 fue otorgado a David Baker por el desarrollo de Ros para el diseño computacional de proteínas, y a Demis Hassabis y John Jumper por el desarrollo de AF3. Ambos, Ros y AF3, usan herramientas de inteligencia artificial para realizar predicciones precisas de estructuras proteicas.
Con base en la teoría clásica de la actividad óptica, se desarrolló un modelo de predicción de espectros de DC a partir de la estructura 3D. Este modelo se fundamenta en el análisis de una base de espectros de alta resolución obtenida del repositorio Protein Circular Dichroism Data Bank (PCDDB) [5], junto con sus correspondientes estructuras del PDB. En un artículo publicado recientemente [6], se describen las principales características de dicho modelo, además de los aspectos generales para la implementación de un servidor público de predicción de espectros, análisis del contenido de estructuras secundarias (porcentaje de hélices alfa, hojas beta, configuraciones desordenadas y otras estructuras), así como la posibilidad de comparación con espectros experimentales. El servidor denominado KCD está disponible de forma gratuita y accesible en el sitio web: https://kcd.cinvestav.mx, el cual ya cuenta con múltiples usuarios de diversos países.
Cabe mencionar que existen métodos precisos de determinación de la estructura 3D de proteínas, como criomicroscopía electrónica, difracción de rayos-X y resonancia magnética nuclear. Sin embargo, estos pueden ser lentos y requerir de condiciones especiales como la preparación y congelación de la muestra en estudio. Frecuentemente, un cambio conformacional de una proteína puede verse reflejado en una modificación de su espectro de DC. El método KCD es capaz de estimar teóricamente dicho cambio, por ejemplo, a partir de los resultados de una simulación de dinámica molecular. Esto permite la comparación entre los espectros experimentales y calculados, para poder tener una mejor idea de las posibles modificaciones estructurales. A continuación, se describe otro uso del servidor.
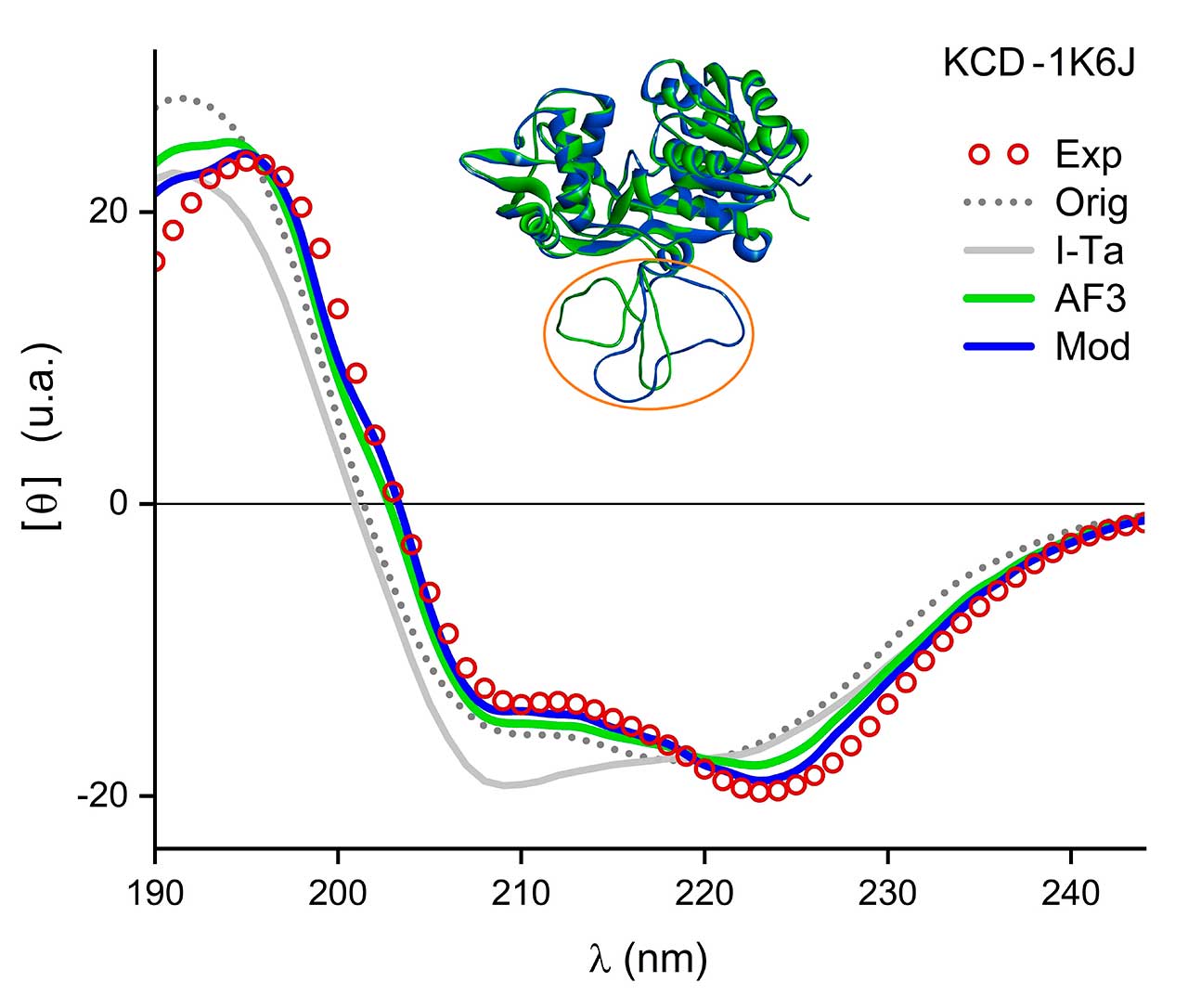
En la Figura 1 se presenta un ejemplo de aplicación del método KCD, para el caso de la reconstrucción de residuos faltantes para un dominio de la proteína NmrA (nitrogen metabolic regulation A), cuyo identificador del PDB es 1K6J. Cabe mencionar que, por diversas razones, no siempre es posible resolver espacialmente la totalidad de los residuos de ciertas proteínas, como en el caso de la proteína NmrA, la cual tiene un faltante de 31 residuos (del 288 al 314) de un total de 353 residuos en cada uno de sus dos dominios o cadenas. Para modelar los residuos ausentes, se emplearon los algoritmos I-Ta [3], AF3 [2] y Modeller (Mod) [7]. En la Figura 1 se muestra la comparación del espectro experimental del PCDDB [5] junto con cuatro espectros teóricos obtenidos a través del método KCD para los casos de la proteína incompleta (Orig), así como los espectros de las proteínas reconstruidas mediante los algoritmos I-Ta, AF3 y Mod. En la Figura también se muestran las dos mejores reconstrucciones 3D, resaltando la región reconstruida. Haciendo uso de una métrica de cercanía basada en las desviaciones absolutas normalizadas entre los espectros teórico y experimental [6], se obtienen los siguientes resultados: 0.29 Orig, 0.30 I-Ta, 0.16 AF3 y 0.09 Mod, lo que apunta a una mejor reconstrucción por medio del algoritmo Mod, que conserva las coordenadas de la estructura de partida y sólo agrega los residuos faltantes a la estructura existente. Es importante señalar que la predicción de AF3 se hace a partir de la secuencia primaria de aminoácidos de la proteína y sin hacer uso de ninguna otra información estructural adicional y, por lo tanto, se puede hacer el ejercicio de especular que el método AF3 requiere de más información para mejorar el modelado de las regiones desestructuradas. Los resultados de esta Figura reflejan el valor de una comparación espectral rápida. Otras aplicaciones del método KCD pueden ser consultadas en la referencia [6].
Referencias
[1] Berman, H. M. et al. (2000). The protein data bank. Nucleic Acids Research, 28 (1), 235-242. https://doi.org/10.1093/nar/28.1.235.
[2] Abramson, J. et al. (2024). Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature, 630, 493-500. https://doi.org/10.1038/s41586-024-07487-w.
[3] Yang, J. y Zhang, Y. (2015). Protein structure and function prediction using I-TASSER. Current Protocols in Bioinformatics, 52, 5.8.1–5.8.15. https://doi.org/10.1002/0471250953.bi0508s52.
[4] Baek, M. et al. (2021) Accurate prediction of protein structures and interactions using a three-track neural network. Science, 373, 871-876. https://doi.org/10.1126/science.abj8754.
[5] Ramalli, S. G., Miles, A. J., Janes, R. W. y Wallace, B. A. (2022). The PCDDB (Protein Circular Dichroism Data Bank): A bioinformatics resource for protein characterisations and methods development. Journal of Molecular Biology, 434, 167441–8. https://doi.org/10.1016/j.jmb.2022.167441.
[6] Jacinto-Méndez, D., Granados-Ramírez, C. G. y Carbajal-Tinoco, M. D. (2024) KCD: A prediction web server of knowledge-based circular dichroism. Protein Science, 33(4), e4967-12. https://doi.org/10.1002/pro.4967.
[7] Webb, B. y Sali, A. (2016). Comparative protein structure modeling using MODELLER. Current Protocols in Bioinformatics, 54, 5.6.1-5.6.37. https://doi.org/10.1002/cpbi.3.