Una herramienta que dejó huella: 30 años de marcadores moleculares en plantas en el Cinvestav
El análisis genético depende de la capacidad de detectar diferencias (polimorfismos) entre individuos. Mendel por ejemplo, aprovechó las características visibles como tamaño, forma, color, entre otros rasgos (1). Más tarde algunos investigadores emplearon marcadores bioquímicos como proteínas u otros metabolitos como indicadores genéticos (2). Sin embargo, la mayoría de los caracteres estudiados se basó en mutaciones con efectos deletéreos, dificultando la posibilidad de dar seguimiento a varios caracteres que se presentan de manera simultánea.
Fundado en los avances en la metodología para manipular y visualizar fragmentos de ADN, Botstein (3) propuso un método apoyado en la detección de polimorfismos en la longitud de fragmentos de restricción, RFLP por sus siglas en inglés. Los RFLP se basan en diferencias en la capacidad de una enzima de restricción para reconocer y recortar una secuencia específica en el ADN. Este método tiene la ventaja que es neutral (no afecta físicamente al organismo del cual se extrae la muestra), y se sustenta directamente en el genotipo, evitando efectos ambientales o de la etapa de desarrollo del organismo, en la detección de los polimorfismos. A diferencia de un marcador fenotípico, como un marcador bioquímico, que tendría enormes variaciones dependiendo del tejido, etapa de desarrollo, y condiciones ambientales en los cuales se toma la muestra. Además, una misma metodología puede aplicarse a cualquier organismo (Figura 1). El desarrollo de los RFLP y un poco más tarde, los análisis de microsatélites o SSR (método basado en diferencias en el número de repeticiones de secuencias específicas de ADN, y empleado hasta la fecha para la identificación de individuos en casos criminales, de paternidad, etcétera), abrió la posibilidad de ampliar el análisis genético a organismos fuera de los modelos de estudio preferidos: maíz, ratón, Drosophila y empezar a correlacionar fenotipos con genotipos.

Sin embargo, la metodología para detectar los polimorfismos RFLP o SSR es complicada, tardada y resulta difícil analizar varios marcadores simultáneamente. Con la innovación de la metodología de la reacción en cadena de la polimerasa (PCR), se buscó la manera de aprovechar la PCR para detectar polimorfismos (4). El primer método desarrollado basado en PCR, fue el de ADN polimórfico amplificado al azar (RAPD) que, aunque fue utilizado ampliamente, es difícil de replicar entre diferentes laboratorios (5).
En 1993 apareció una solicitud de patente por parte de la empresa Keygene establecida en Holanda para una nueva metodología combinando la detección de polimorfismos en sitios de restricción (igual que los RFLPs) con el uso de PCR (igual que los RAPD). Este procedimiento se conoce como polimorfismos en la longitud de fragmentos amplificados (AFLP), y además de las ventajas que ofrecen los RFLP, SSR y RAPD, la metodología de AFLP es robusta y facilita el análisis de cientos de marcadores a través del genoma de manera simultánea (6,7).
El método de AFLP se basa en la reducción del número de fragmentos de restricción que serán analizados. Si simplemente cortamos el ADN de un organismo con una enzima de restricción, y separamos los fragmentos por electroforesis en un gel, observaremos un barrido continuo de los cientos de fragmentos sin poder distinguir los fragmentos individuales, y aunque existen diferencias en la presencia de sitios de restricción entre distintos individuos, es imposible detectar estos polimorfismos. La metodología de AFLP permite la visualización de fragmentos individuales y su comparación entre diferentes individuos de manera sencilla.
Típicamente, el proceso experimental para los AFLPs se inicia con la digestión del ADN genómico del individuo usando dos enzimas de restricción tipo II distintas: una enzima que reconoce un sitio de seis pares de bases y otra enzima que detecta un sitio de cuatro pares de bases. En el siguiente paso se agregan adaptadores diseñados para unir específicamente a los sitios de restricción cortados por las enzimas y se lleva a cabo una reacción de PCR empleando oligonucleótidos con homología específica para los adaptadores (Figura 2). Dadas las condiciones de la reacción, los fragmentos con sitios de restricción para ambas enzimas son preferencialmente amplificados, mientras los fragmentos con sitios de restricción para una sola enzima no se amplifican de manera óptima. Así, se enriquece la muestra para los fragmentos con extremos portando sitios para diferentes enzimas. Sobre esta reacción enriquecida se lleva a cabo una segunda amplificación, con oligonucleótidos selectivos: los mismos que se utilizaron en la primera ronda de amplificación, pero ahora añadiendo 1 a 3 nucleótidos al azar extras hacia los extremos 3´ del fragmento de interés. De esta manera solamente los fragmentos que portan estos nucleótidos enseguida del sitio de restricción, serán amplificados, reduciendo de nuevo la complejidad del número de fragmentos y permitiendo su visualización después de su separación por electroforesis. Por cada nucleótido selectivo se reduce alrededor de 4 veces el número de fragmentos que serán detectados. La incorporación de nucleótidos marcados (por radiactividad o fluorescencia) en uno de los oligonucleótidos permite la visualización de los fragmentos de manera óptima (Figura 2).
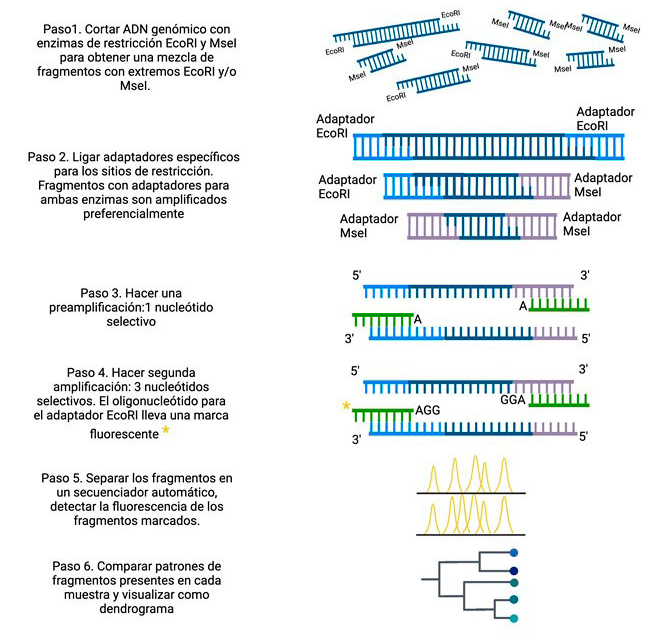
Es posible modificar la metodología de los AFLP para acomodar genomas pequeños como los de bacterias, reduciendo a 1 el número de nucleótidos selectivos o aumentándolos a 3 cuando el análisis involucra especies con genomas grandes como en el género Agave. Generalmente se amplifican entre 50 y 100 fragmentos, los cuales se separan por electroforesis en geles de acrilamida o en un secuenciador automático “tradicional” (secuenciación Sanger), donde se puede resolver fragmentos con diferencias de un solo nucleótido.
La Unidad Irapuato del Cinvestav fue el primer sitio en México donde se implementaron los métodos de análisis de marcadores moleculares tipo RFLP, RAPD y SSR en especies vegetales, específicamente en maíz y frijol, y el siguiente paso fue la incorporación de la metodología de los AFLP. Los colegas de Keygene generosamente permitieron acceso a la aplicación para la patente de los AFLP antes de su publicación formal (7), y con la infraestructura para la secuenciación de ADN ya existente en el Departamento de Ingeniería Genética de la Unidad Irapuato, fue relativamente sencillo instrumentar la metodología y atender el interés creciente para llevar a cabo trabajos de marcadores moleculares en plantas. La ubicuidad del método permitió aplicarlo a 60 especies vegetales, pero también en una amplia variedad de especies y géneros fuera del campo vegetal, incluyendo 11 especies de hongos, 3 de bacterias, 1 insecto y 1 especie de coral. La metodología de AFLP sentó las bases para un gran número de publicaciones y tesis de Licenciatura, Maestría y Doctorado del grupo de Genética Molecular de la Unidad Irapuato, que hasta la fecha ofrece servicios internos y externos de análisis de marcadores genético-moleculares incluyendo AFLP.
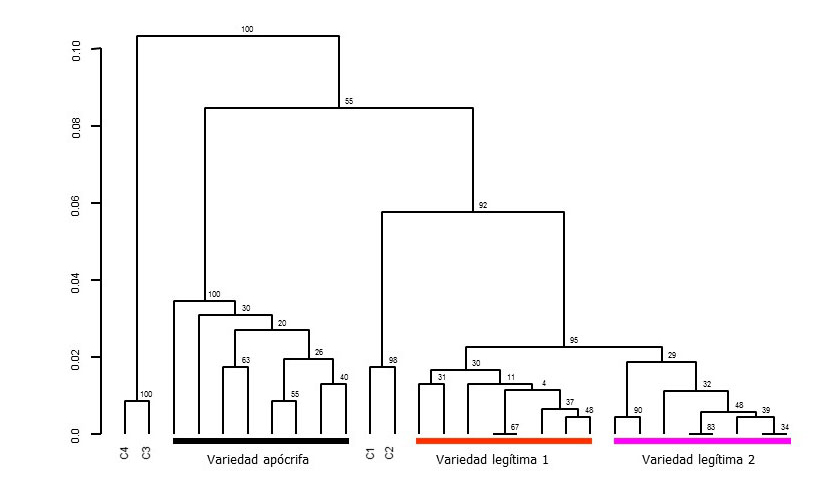
Las aplicaciones incluyen estudios de diversidad, por ejemplo en agave, piña y limón, entre otros; el desarrollo de mapas genéticos y asociación con caracteres de interés en maíz, frijol y varias especies de hongos y análisis de poblaciones de hongos patógenos. Algunos trabajos externos se han apoyado en la certificación de variedades y en resolver disputas de propiedad a nivel judicial con la determinación de diferencias o similitudes entre variedades comerciales de maíz y jitomate, producidos por distintas empresas de semillas. En casos de demandas específicamente con semillas de chile y melón, fue posible mostrar de manera inequívoca la venta de semillas apócrifas (Figura 3).
En colaboración con el Dr. José Ruíz Herrera (Q.E.P.D.), cuyo interés se enfocó en los cambios en patrones de metilación durante el dimorfismo en hongos, fue posible desarrollar una aplicación novedosa para los AFLP. Inicialmente los análisis de metilación estaban dirigidos hacia genes individuales, un trabajo laborioso que necesitaba conocimiento de las secuencias de los genes o regiones de interés del genoma. Adaptando la metodología de AFLP e incorporando enzimas de restricción sensibles a la presencia de nucleótidos metilados, fue posible extender la metodología de los AFLP para estudiar cambios en la metilación a través del genoma completo. Esta metodología se aplicó a estudiar cambios durante las diferentes etapas del dimorfismo de Mucor rouxii, Yarrowia lipolytica y Ustilago maydis (8). Es así como nació la metodología después conocida como MSAP-polimorfismos amplificados sensibles a la metilación (9). Aunque fue desarrollado originalmente en hongos, MSAP ha sido aprovechado ampliamente en especies vegetales, en Drosophila, y en mamíferos como humanos y ratón, entre otros (10).
Si bien AFLP y MSAP son utilizados ampliamente todavía (11, 12), la tendencia es reemplazar estas metodologías con nuevas estrategias para genotipificación basada en la Secuenciación de Nueva Generación (NGS). La reducción en costos para la secuenciación de genomas completos y el desarrollo de la metodología de “Genotyping by Sequencing o GBS”, basada en polimorfismos en nucleótidos individuales (single nucleotide polymorphisms), hace que la metodología de AFLP/MSAP ya no sea competitiva. En un sentido, el método de AFLP fue víctima de su propio éxito dado que los métodos de GBS emplean la misma estrategia de digestión con enzimas de restricción y amplificación de un número reducido de fragmentos, los cuales son secuenciados en lugar de ser separados por electroforesis (13, 14).
La historia de los AFLPs es un excelente ejemplo de cómo resolver un problema (reducir la cantidad de ADN para genotipificar) con una idea relativamente sencilla. Esta idea “sencilla” ha sido extendida para diferentes propósitos y ha tenido un impacto significativo con la publicación de por lo menos 300,000 artículos en revistas internacionales. Aunque el método original sin duda será obsoleto en los próximos años, el incremento acelerado en el uso de GBS asegura que la esencia del método de AFLP se mantendrá vigente al menos otros 30 años.
Referencias:
- Mendel G. (1866). Versuche über Pflanzen-Hybriden Verhandlungen des naturforschenden Vereines in Brünn, 4, 3-47.
- Brücher, H. (1989). Protein Plants. In: Useful Plants of Neotropical Origin. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-73313-0_4
- Botstein, D., White, R.L., Skolnick, M., Davis, R.W. (1980). Construction of a genetic linkage map in man using restriction fragment length polymorphisms. American journal of human genetics, 32(3), 314-331.
- Mullis, K.B., Faloona, F.A. (1987). Specific synthesis of DNA in vitro via a polymerase-catalyzed chain reaction. In Methods in enzymology Vol. 155, 335-350. Academic Press.
- Williams J.G.K., Kubelik A.R., Livak K.J., Rafalski J.A., Tingey S.V., (1990). DNA polymorphisms amplified by arbitrary primers are useful as genetic markers. Nucleic Acids Res, 18, 6531-6535.
- Vos P., Hogers R., Bleeker M., Reijans M., Van de Lee T., Hornes M., Fijters A., Pot J., Peleman J., Kuiper M., Zabeau M. (1995). AFLP: a new technique for DNA fingerprinting. Nucleic Acids Res 23:4407–4414.
- Zabeau M., Vos P. (1993). Selective restriction fragment amplification: a general method for DNA fingerprinting. European Patent Application number: 92402629.7, Publication number 0534 858 A1.
- Reyna-López, G.E. Simpson J., Ruiz-Herrera J. 1997 Differences in DNA methylation patterns are detectable during the dimorphic transition of fungi by amplification of restriction polymorphisms. Mol. Gen. Genet. 16, 847-852.
- Zhao Y., Chen M., Storey K. B., Sun L., Yang H. (2015). DNA methylation levels analysis in four tissues of sea cucumber Apostichopus japonicus based on fluorescence-labeled methylation-sensitive amplified polymorphism (F-MSAP) during aestivation. Comparative Biochemistry and Physiology Part B: Biochemistry and Molecular Biology, 181, 26-32.
- Fulneček J., Kovařík A. (2014). How to interpret methylation sensitive amplified polymorphism (MSAP) profiles?. BMC genetics, 15(1), 1-9.
- Panagiotopoulou H., Marzecki K., Gawor J., Kuhl H., Koper M., Weglenski P.,& Rzepkowska M. (2023). Extensive search of genetic sex markers in Siberian (Acipenser baerii) and Atlantic (A. oxyrinchus) sturgeons. Aquaculture, 573, 739517. 1-11.
- Vogt G. (2023). Evolution, functions and dynamics of epigenetic mechanisms in animals. In Handbook of Epigenetics. Academic Press. 521-549.
- Deschamps S., Llaca V., May G. D. (2012). Genotyping-by-Sequencing in Plants. Biology, 1(3), 460-483.
- He J., Zhao X., Laroche A., Lu Z.-X., Liu H., Li, Z. (2014). Genotyping-by-sequencing (GBS), an ultimate marker-assisted selection (MAS) tool to accelerate plant breeding. Frontiers in Plant Science, 5, 484. 1-8.
- BioRender.com https://app.biorender.com/biorender-templates Subscription: Institution, Agreement number: GL25XHLEAV, Journal name: Avance y Perspectiva.