El Premio Nobel de Química 2024 se otorgó al diseño computacional y la predicción de la estructura de proteínas por medio de AlphaFold, herramienta que revolucionó la determinación de estructuras proteicas. La historia de AlphaFold comienza en la compañía DeepMind, con el desarrollo del famoso programa de inteligencia artificial que se conoce como AlphaGo que venció a los mejores jugadores humanos de Go. La victoria en el campo del juego fue sólo el comienzo: los principios que permitieron el éxito de AlphaGo se aplicaron para enfrentar un reto mayor: el plegamiento de proteínas.

Las proteínas son macromoléculas fundamentales para el funcionamiento de los seres vivos; son estructuras poliméricas de grandes dimensiones, tanto en extensión espacial como en peso molecular, y se forman mediante la unión de unidades básicas —los aminoácidos—, a través del llamado enlace peptídico. Estas estructuras se conocen como cadenas polipeptídicas. Existen proteínas de una y de dos o más cadenas polipeptídicas.
Las proteínas tienen cuatro niveles de descripción estructural. La primaria es la secuencia de aminoácidos que la constituyen; la secundaria surge como consecuencia del rearreglo de la columna vertebral de la proteína para formar hélices ? y hojas ?, principalmente; la terciaria es la estructura tridimensional que adopta una proteína como consecuencia de las interacciones no covalentes (puentes de hidrógeno, interacciones electrostáticas y de dispersión) entre los grupos laterales de los aminoácidos. Finalmente, las proteínas que tienen más de una cadena polipeptídica se ensamblan para dar lugar a la estructura cuaternaria.
La actividad esencial de una proteína se manifiesta cuando adquiere una estructura tridimensional o conformación particular que se conoce como estructura nativa. El paso de una proteína desde su secuencia de aminoácidos hasta la estructura nativa era uno de los grandes retos de la bioquímica, la biología molecular y fisicoquímica de proteínas. A este problema se le conoce como plegamiento, y comenzó a estudiarse desde los años sesenta del siglo XX. Se abordó macroscópicamente con herramientas termodinámicas y cinéticas, y también microscópicamente con el propósito de entender y reconocer cuáles son las interacciones dominantes entre los aminoácidos de una proteína que dirigen a una cadena peptídica a adquirir su estructura nativa.
A principios de los años 60 del siglo pasado, un concepto muy arraigado en la estructura y función de las proteínas era el papel de los llamados puentes disulfuro como determinantes para la estabilidad y el plegamiento que llevaba a una proteína a adquirir su estructura nativa. En 1973, Christian B. Anfinsen, químico y Premio Nobel de Química en 1972, propuso una idea innovadora. Su hipótesis termodinámica establece que la estructura nativa de una proteína es la termodinámicamente más estable, esto es, la que tiene la menor energía libre, y depende de la secuencia de aminoácidos —estructura primaria— y del medio en la que se encuentra, más no en factores cinéticos.
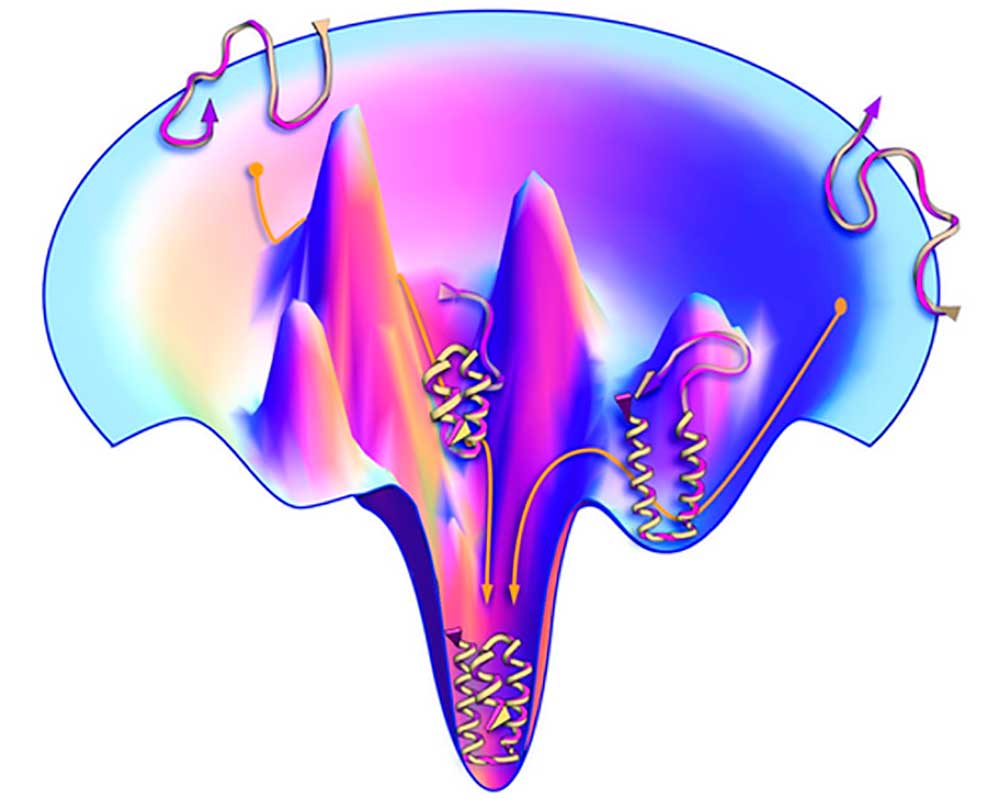
La complejidad del problema queda patente con la paradoja de Levinthal quien en 1969 señaló que el número de posibles arreglos conformacionales de una proteína es tan estratosfericamente alto que transitar al estado nativo no puede hacerse por caminos estocásticos, al azar, pues éstos tomarían un tiempo mayor a la edad del Universo. La paradoja de Levinthal y la hipótesis termodinámica de Anfinsen parecen estar en conflicto, el cual se supera con la teoría del paisaje molecular (molecular landscape) y del embudo, propuesta por Peter Wolynes y Ken Dill a mediados de los años 90 del siglo XX. En la Figura 1 se muestra el mecanismo de plegamiento de acuerdo con la teoría del paisaje molecular y el embudo.
El reto histórico: La revolución de AlphaFold
El pionero en el uso de herramientas informáticas para la predicción de estructuras proteicas fue David Baker, uno de los ganadores del Premio Nobel de Química 2024. Hace 20 años, Baker y su equipo desarrollaron un programa, que llamaron Rosetta, para la predicción de estructura de proteínas a partir de principios físicos, como los conceptos desarrollados por Wolynes y Dill, y algoritmos computacionales. Antes del advenimiento de AlphaFold, Rosetta era la herramienta más confiable y de primera elección para predecir la estructura proteica.
Esta situación cambió drasticamente al conocerse los resultados de AlphaFold en 2018. A AlphaFold lo desarrolló DeepMind, ahora parte de Google, y demostró ser capaz de predecir la estructura de proteínas con una precisión sorprendente, en muchos casos igualando la calidad de los métodos experimentales tradicionales, pero de forma más rápida y accesible. No es posible entender el asombroso éxito de AlphaFold sin la existencia del enorme proyecto Protein Data Bank (PDB), un repositorio de estructuras de proteínas obtenidas por cristalografía de rayos-X, criomicroscopía electrónica y resonancia magnética nuclear.
AlphaFold ha pasado por varias versiones que han mejorado continuamente su capacidad de predicción. El debut de AlphaFold ocurrió en 2018 y se destacó al predecir estructuras de algunas proteínas difíciles. En su presentación, ganó el concurso bianual de predicción de estructuras de proteínas conocido como CASP, siglas en inglés de Evaluación Crítica de Técnicas para la Predicción Estructural de Proteínas. Poco después, en 2020, AlphaFold2 fue lanzado; representó un salto revolucionario, alcanzando una precisión sin precedentes para proteínas individuales que le valió el Premio Nobel. AlphaFold sigue mejorando, y con las versiones más recientes no sólo predice estructuras de proteínas individuales, sino también de complejos proteicos con ADN, ARN y diversos ligandos e iones.
Aplicaciones prácticas
AlphaFold ya tiene un impacto importante en diversas áreas de estudio. En nuestro grupo de investigación hemos aplicado AlphaFold para predecir la estructura de proteínas que contienen metales, un área donde esta tecnología aún presenta muchos desafíos. Aunque AlphaFold no incluye explícitamente información sobre cómo los aminoácidos interactúan con los metales, hemos encontrado que las predicciones son notablemente cercanas a la evidencia experimental.
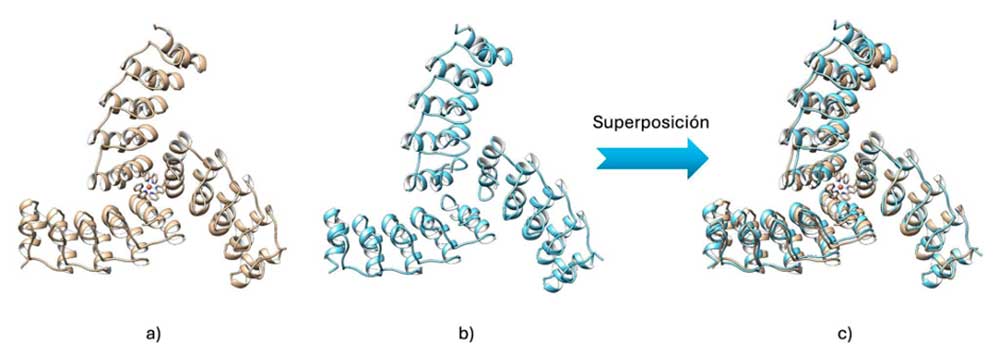
Investigadores de todo el mundo utilizan esta herramienta en proyectos innovadores. Se ha empleado para estudiar proteínas implicadas en enfermedades como el Alzheimer, identificar nuevas rutas metabólicas en bacterias, y ayudar al diseño de proteínas sintéticas con funciones completamente nuevas.
El futuro de AlphaFold
AlphaFold promete ser una herramienta crucial en el avance de la biomedicina, la biotecnología y muchas otras disciplinas científicas en los años venideros.
Los logros alcanzados en la predicción de la estructura de proteínas con Rosetta y AlphaFold son espectaculares. Sin embargo, todavía hay muchos desafíos. Por ejemplo, mejorar la descripción dinámica de proteínas, predecir interacciones con membranas celulares y garantizar un acceso equitativo a esta tecnología en todo el mundo, son algunas de las áreas de investigación en esta disciplina que requerirán atención en el futuro.
El Premio Nobel de Química 2024 no solo reconoce el avance técnico de la inteligencia artificial, sino que marca el inicio de una nueva era en la ciencia, donde la ciencia y las herramientas informáticas están más entrelazadas que nunca.
Referencias
https://www.nobelprize.org/all-nobel-prizes-2024/
https://www.nobelprize.org/prizes/chemistry/1972/press-release/
https://es.wikipedia.org/wiki/AlphaFold
Honig, B.. Protein folding: from the levinthal paradox to structure prediction. Journal of Molecular Biology, 293, 283-293 (1999).
Jumper, J., Evans, R., Pritzel, A. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021)
Ken A. Dill Theory for the folding and stability of globular proteins. Biochemistry 24, 1501-1509 (1985)
Hans Frauenfelder et al. The Energy Landscapes and Motions of Proteins. Science 254, 1598-1603(1991)
Imagen de portada : David Baker, Demis Hassabis y John M. Jumper. Premio Nobel de Química 2024. Ilustraciones de Niklas Elmehed. Divulgación del Premio Nobel