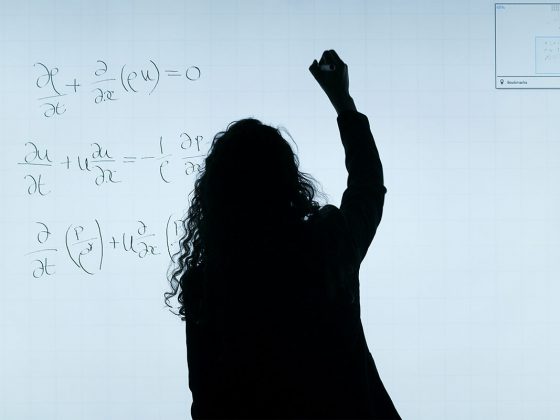La biología cuenta con un gran número de herramientas para estudiar a los organismos vivos. Existen aquellas que, de manera tradicional, tratan de responder los cuestionamientos desde una visión individual del problema, a través de la generación de una hipótesis; también contamos con otras que, combinadas, tratan de explicar los sistemas como un todo. A la integración de estas herramientas se le conoce como biología sistémica, la cual se respalda en estudios conocidos como ciencias ómicas, muy utilizados en la actualidad en el ámbito mundial y donde, a través de un acercamiento holístico, se construyen las hipótesis a probar a partir de la generación y análisis previo a los datos.
Las ciencias ómicas siguen un camino bidireccional desde el estudio de los genes en el ADN (genómica), pasando por el análisis estructural y funcional de las proteínas (proteómica) hasta la metabolómica, la cual se encarga de identificar y cuantificar los compuestos de bajo peso molecular derivados de los procesos biológicos en los organismos. A estos compuestos se les conoce como metabolitos. Su presencia, concentración y flujo resulta de un complejo entramado entre la expresión génica, las proteínas y el ambiente.
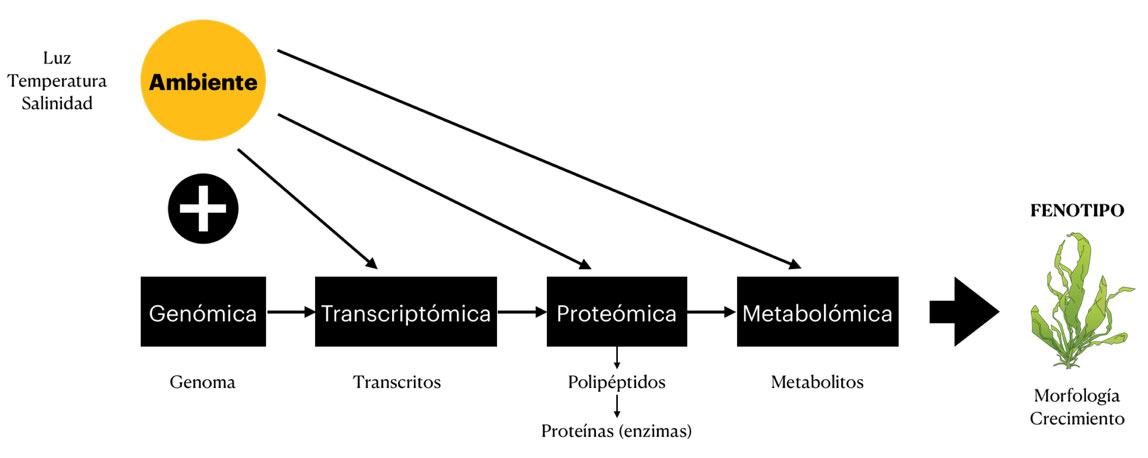
Y es aquí donde entra en juego la transcriptómica. Esta ómica nos permite analizar las variaciones en los transcritos (códigos de ARN), de los organismos cuando se exponen a condiciones ambientales concretas. Éstos se traducen en proteínas que se encargan de la mayoría de los procesos biológicos y que terminarán definiendo el fenotipo del ente biológico. En pocas palabras, la transcriptómica nos permite tomar una fotografía de un momento concreto para entender cómo el ambiente, en conjunción con el genotipo, modelan su apariencia, permitiéndonos identificar aquellos genes que se activan como consecuencia de estímulos específicos y dan como resultado condiciones fisiológicas concretas (Figura 1).
Hoy en día, la tecnología nos permite observar más de cerca estos procesos, siendo la secuenciación de ARN (RNA-seq, por sus siglas en inglés) la más popular entre los estudiosos del transcriptoma, principalmente por su rapidez, robustez y su costo cada vez más accesible.
La RNA-seq es una herramienta en constante evolución. Ofrece ventajas claves con respecto a otras técnicas muy usadas en la secuenciación de ARN como los microarreglos; por ejemplo, la cantidad mínima de ARN que requiere, incluso no es necesario conocer la secuencia genómica per se, por lo que la convierte en una tecnología atractiva para el estudio de organismos no modelo. Además, la RNA-seq tiene altos niveles de reproducibilidad, tanto en réplicas técnicas como biológicas, lo que permite capturar de manera fidedigna la dinámica del transcriptoma entre dos muestras o tratamientos. Las operaciones llevadas a cabo en estos análisis son repetitivas y se benefician de la computación, por lo tanto, un hardware promedio puede ayudarnos a analizar la información obtenida si se trata de experimentos “sencillos”. Sin embargo, la gran cantidad de información generada por la RNA-seq y la complejidad del transcriptoma representan algunos de los retos al momento de interpretar los resultados, donde la bioinformática nos asiste en los estudios transcriptómicos.
Finalmente, y con el objetivo de validar el análisis, se cuantifica la expresión génica en los genes de interés usando técnicas como la reacción en cadena de la polimerasa (PCR, por sus siglas en inglés), en tiempo real o cuantitativa, un método común y estadísticamente medible. No obstante, para obtener mejores resultados, es importante considerar la validación funcional de estos genes en la planeación previa de nuestros experimentos. Adicionalmente, los patrones de expresión génica deben estar ligados a los cambios observados en el fenotipo en la fase de experimentación.
Y es así, como si fuera una potente lupa, que toda la información obtenida de esta ómica a través de la RNA-seq nos proporciona una oportunidad para aumentar nuestra comprensión sobre los genes involucrados en los procesos fisiológicos o rutas de síntesis de compuestos de interés en organismos con un alto valor comercial; por ejemplo, las algas marinas.
Macroalgas marinas: más allá de su importancia ecológica
Además de ser responsables de oxigenar nuestro planeta, la importancia ecológica de las algas marinas es indiscutible, no sólo como agentes constructores de hábitats y de protección contra la erosión, sino también por su contribución al secuestro de carbono y la remoción de nutrientes del océano. Las macroalgas son algas multicelulares, macroscópicas, y con una gran diversidad morfológica. Existen aproximadamente 25,000 especies descritas hasta ahora, clasificadas con base en sus pigmentos y coloración en Phaeophyceae, Chlorophyta y Rhodophyta (algas cafés, verdes y rojas, respectivamente). Viven en un medio muy cambiante y complejo. Constantemente están sujetas a variaciones en la luz que reciben, la temperatura, salinidad, nutrientes, oleaje y mareas. Es así que, como forma de adaptación, han desarrollado una gran capacidad para producir numerosos compuestos y metabolitos, muchas veces únicos y de gran interés para el hombre.
En los últimos años, el mercado y consumo de macroalgas ha aumentado exponencialmente en el mundo, siendo los países asiáticos los principales productores y consumidores. Además de contar con un alto valor nutricional, las macroalgas producen compuestos como pigmentos, fenoles, hidrocoloides y micosporinas, con propiedades antioxidantes, antimutagénicas, anticoagulantes, anticancerígenas y antibacteriales, por lo que son utilizados como agroquímicos, cosméticos, biomateriales, bioenergéticos, medicamentos y alimentos [1]. En 2018, según la FAO, el mercado global de macroalgas tenía un valor de aproximadamente $13 billones de dólares estadunidenses [2] y a pesar del gran número de especies conocidas, actualmente el 98% de la producción (extracción y cultivo) está comprendida en cinco géneros: Pyropia, Undaria, Gracilaria, Kappaphycus y Saccharina [3].
Hasta la fecha se ha publicado una cantidad importante de estudios transcriptómicos en algas; sin embargo, sólo se han obtenido los genomas completos de muy pocas especies. Mediante los estudios del transcriptoma se han decodificado sus genes, sus redes regulatorias, y se ha profundizado en la comprensión de la relación metabolito- gen, lo que ha permitido diseñar proyectos más contundentes en los campos de la ingeniería metabólica y la biología sintética con el objetivo de identificar macroalgas productoras de compuestos con interés comercial. No obstante, otros trabajos se han enfocado en estudios ecológicos y en la investigación del microbioma asociado, logrando identificar proteínas relacionadas con comunidades microbianas que participan activamente en la morfogénesis y el crecimiento del alga. Así, los avances en las tecnologías de secuenciación en conjunto con la disminución de sus costos han permitido una expansión notable en el análisis transcriptómico en estos organismos. Sin embargo, y a pesar de la gran cantidad de trabajos realizados en el campo de la fisiología algal, aún se desconocen, o no se comprenden del todo, algunos mecanismos moleculares relacionados con la capacidad de las algas para adaptarse a condiciones de estrés, y por lo tanto de producir los metabolitos de interés. Al ser organismos que habitan ambientes muy dinámicos, las macroalgas son las candidatas idóneas para desarrollar investigación sobre el estrés abiótico, por lo que incluir estudios del transcriptoma en la investigación fisiológica algal permitiría dilucidar la expresión génica en condiciones ambientales específicas o de cultivo.
Historia transcriptómica en macroalgas
Diversos estudios recientes, han evaluado la expresión de los genes con interés económico en macroalgas (Figura 2). La mayor cantidad de trabajos transcriptómicos generados hasta la fecha se enfocan en el estudio de algas cafés, seguido de las rojas y por último en algas verdes. El interés en cada grupo radica en su importancia económica, su capacidad de sintetizar compuestos bioactivos, su importancia ecológica y en la rápida expansión mundial de su cultivo y/o extracción comercial.
El primer análisis transcriptómico data del año 2000, utilizando como modelo el alga café Laminaria digitata, comúnmente llamada “alga kombu” y que hace las delicias de los aficionados a la comida japonesa. Se trabajó con una técnica de secuenciación de primera generación llamada Sanger, que actualmente se emplea muy poco [4]. Siete años más tarde, se reporta uno de los primeros trabajos con algas rojas, en particular con Chondrus crispus, o “musgo irlandés”, macroalga apreciada y valorada por las propiedades de su polisacárido en las industrias alimenticia y farmacéutica. En este trabajo se utilizó la técnica de microarreglos [5]. No es hasta 2011 cuando se utiliza el análisis transcriptómico para el estudio de un alga verde, Ulva pertusa, especie que arriba masivamente a las costas de China y es apreciada por su valor nutricional [6]. Este mismo año se populariza la tecnología de secuenciación de segunda generación con la plataforma Illumina en estudios de macroalgas, a pesar de que esta herramienta surge alrededor de 2006.
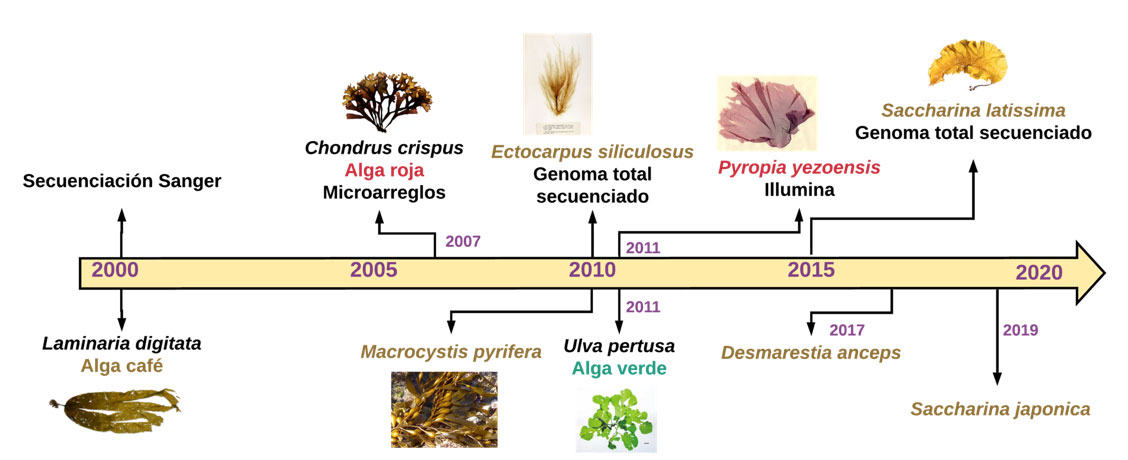
A la fecha, los tres géneros más estudiados son Saccharina (alga café), y las algas rojas Pyropia (alga nori) y Gracilaria, todas ellas productoras de polisacáridos y con una demanda significativa en la industria alimenticia en países asiáticos. La mayoría de los artículos publicados responden a estudios descriptivos enfocados a estudiar procesos fisiológicos en el ámbito molecular con el objetivo de entender procesos biológicos de adaptación o aclimatación; otra gran parte de la investigación se centra en estudios ecológicos en organismos silvestres, y sólo pocos estudios corresponden a ensayos para optimizar el crecimiento de macroalgas para su cultivo comercial o en identificación taxonómica. Los análisis relacionados con el cultivo han permitido identificar la respuesta de las macroalgas a parámetros específicos con el objetivo de seleccionar cepas con un mayor crecimiento y rendimiento en la producción de metabolitos de interés. Además, los datos obtenidos del análisis transcriptómico ayudan a construir filogenias, medir selección natural e incluso entender la evolución de los genes entre especies de macroalgas o plantas terrestres.
Un ejemplo: en 2015, se generó un análisis para bosquejar el genoma de Saccharina japonica en diferentes cultivos en China. La RNA-seq resultó en una secuencia genómica de 537 millones de pares de bases, que corresponde al 98.5% del genoma estimado, además de que fueron anotados 18,733 genes codificantes. Estos genes están vinculados con la síntesis de la pared celular (relacionada con sus polisacáridos), concentración de compuestos halogenados, y con procesos del desarrollo. Adicionalmente, fueron secuenciados siete individuos de cultivo y nueve silvestres, lo que dio a conocer que la diversidad genética en poblaciones silvestres es mayor que en organismos de cultivo. Se identificó que las poblaciones de cultivo son descendientes de un híbrido silvestre entre dos especies diferentes, Saccharina japonica y Saccharina longissima. También fueron registradas variaciones de un solo nucleótido en ambas poblaciones, que pueden utilizarse para la construcción de marcadores que permitan la selección eficiente de cultivares [7].
Presente y Futuro
La integración del análisis transcriptómico como herramienta complementaria en estudios de macroalgas con importancia comercial y ecológica nos permite añadir un mayor número de transcritos, o “códigos” de ARN en las bases de datos públicas (como GenBank) que podrían utilizarse para diseñar investigaciones más completas. A pesar de que se han identificado genes responsivos a condiciones concretas en algunas especies, muy pocos trabajos han complementado esta información con ensayos fisiológicos (como medición de parámetros fotosintéticos) con el crecimiento, o con la producción de metabolitos.
En el futuro, la inclusión de esta información resultará enriquecedora para comprender rutas biológicas específicas y conocer el “fitness” de los organismos, lo cual nos ayudará a seleccionar cepas más productivas y optimizar los protocolos en cultivos acuícolas. Además, la integración de tecnologías de secuenciación masiva más recientes (tercera generación) como PacBio y Oxford Nanopore permiten reconstruir con mayor precisión secuencias largas y hacer el análisis en tiempo real. Aunado a lo anterior, la preparación de muestras toma tan sólo diez minutos, e incluso, se pueden hacer las mediciones in situ por el tamaño tan compacto del instrumento. Adicionalmente, la constante evolución de tecnologías ampliamente utilizadas como Illumina, ya en su versión NovaSeq 6000, presentan un mayor rendimiento y reducen el tiempo de secuenciación. Estas tecnologías ya se emplean en plantas superiores y su uso en el estudio de macroalgas es inminente. Representan un futuro promisorio.

Al aumentar todavía más la capacidad de nuestra “lupa transcriptómica”, herramientas más finas como la secuenciación de una sola célula nos permitirán trabajar con una cantidad mínima de ARN, facilitando la identificación de propiedades celulares complejas, variabilidad entre células e incluso, nuevos tipos celulares, detectando con más eficiencia la variación biológica. Realmente… ¡un futuro prometedor!
La expansión acelerada del cultivo de algas, de sus mercados, y de la necesidad de responder interrogantes sobre su colonización y proliferación en ambientes costeros ha incentivado la generación de estudios transcriptómicos más completos que permiten mejorar su crecimiento, la producción de metabolitos de interés, así como conocer los mecanismos de tolerancia al estrés medioambiental. Sin embargo, la industria acuícola de macroalgas aún no se equipara con la producción comercial de plantas terrestres, por lo tanto, el número de trabajos generados y de secuencias genómicas completas en las primeras son muy inferiores. Tan sólo con buscar en bases de datos especializadas se pueden encontrar decenas de estudios transcriptómicos publicados en 2021 en plantas superiores como Arabidopsis, organismo modelo en fitobiología.
Es de esperar que conforme la producción de macroalgas ascienda y las necesidades del mercado sean mayores, el número de trabajos que incorporen análisis transcriptómicos con el objetivo de optimizar los protocolos de cultivo y aprovechamiento de este recurso aumente. En eso andamos…

Referencias consultadas
- Jayalakshmi, J., Subramanian, V. y Anantharaman, P. (2014). Evaluation of biochemical composition and in vitro antioxidant properties of selected seaweeds from Kanyakumari coast, Tamil Nadu, India. Advances in Applied Science Research, 5(4), 74–81.
- 2020.The State of World Fisheries and Aquaculture 2020. Sustainability in action. Rome.
- Pereira, R. y Yarish, C. (2008). Mass Production of Marine Macroalgae. Encyclopedia of Ecology, (Mayo 2016), 2236–2247.
- Crépineau, F., Roscoe, T., Kaas, R., Kloareg, B. y Boyen, C. (2000). Characterisation of complementary DNAs from the expressed sequence tag analysis of life cycle stages of Laminaria digitata (Phaeophyceae). Plant Molecular Biology, 43(4), 503–513.
- Collén, J., Guisle-Marsollier, I., Léger, J. J. y Boyen, C. (2007). Response of the transcriptome of the intertidal red seaweed Chondrus crispus to controlled and natural stresses. New Phytologist, 176(1), 45–55.
- Fu, W., Shuai, L., Yao, J., Zheng, B., Zhong, M. y Duan, D. (2011). Molecular cloning and expression analysis of a cytosolic Hsp70 gene from Ulva pertusa (Ulvophyceae, Chlorophyta). Journal of Applied Phycology, 23(4), 681–690.
- Ye, N., Zhang, X., Miao, M., Fan, X., Zheng, Y., Xu, D. y Zhao, F. (2015). Saccharina genomes provide novel insight into kelp biology. Nature Communications, 6, 1–11…