Recientemente leímos en las noticias sobre la existencia de nuevas variantes del SARS-CoV-2. Algunas de estas variantes contienen mutaciones que modifican la facilidad con la que el coronavirus se propaga entre las personas y también otras que les permiten evadir parcialmente las defensas inmunológicas del cuerpo, comprometiendo en cierto grado la eficacia de determinadas vacunas. Debido a ello, es fundamental que exista una vigilancia epidemiológica de las variantes que existen en la población. En el Cinvestav hemos implementado una herramienta que permite visualizar el conocimiento que tenemos de la distribución de variantes del SARS-CoV-2 en México. Este instrumento es de libre acceso.
Evolución por divergencia de linajes
Para poder hablar de las variantes del coronavirus, es necesario definir primero qué son y en qué se diferencian una variante de un linaje y de una cepa. Los virus, al igual que los seres vivos, evolucionan. El proceso de evolución consiste en la aparición de variantes genéticas mediante mutación, seguido de herencia diferencial de esas variaciones. Esta transformación (detalles más, detalles menos) fue descrita por Charles Darwin en El Origen de las especies (Figura 1).
El proceso de evolución y divergencia lo podemos imaginar de la siguiente forma. A través de las generaciones, los virus adquieren mutaciones que heredan a sus descendientes. Una variante viral se define por un conjunto específico de mutaciones, las cuales pueden o no cambiar el comportamiento del virus. Con el tiempo, distintas poblaciones virales adquieren diversas mutaciones y comienzan a diferenciarse genéticamente. De esta forma surgen variados linajes que se pueden identificar mediante análisis filogenéticos. Finalmente, cuando se han acumulado suficientes mutaciones, el comportamiento del virus cambia de modo notable. Entonces hablamos de una nueva cepa. Cabe mencionar que en algunas ocasiones bastan pocas mutaciones para cambiar radicalmente la biología de un virus. En el caso del SARS-CoV-2 es muy claro que todas las variantes (y linajes) que existen pertenecen a una misma cepa.

La plataforma Mexstrain
Para visualizar el proceso de evolución que describimos anteriormente, un grupo internacional de investigadores desarrollaron las plataformas Nextstrain (https://nextstrain.org/) y Microreact (https://microreact.org/showcase), que se pueden utilizar para estudiar distintos patógenos y han sido muy importantes para entender la diversidad genética del SARS-CoV-2 en el ámbito global. Si bien nuestro país está representado en la plataforma global de Nextstrain, sólo una fracción pequeña de la diversidad genética del coronavirus en México se muestra con recurrencia en ella. Con el fin de dar seguimiento cercano a la evolución del coronavirus en México, en el Cinvestav hemos implementado una plataforma que denominamos Mexstrain (http://www.ira.cinvestav.mx/ncov.evol.mex.aspx), que utiliza las tecnologías de Nextstrain y de Microreact.
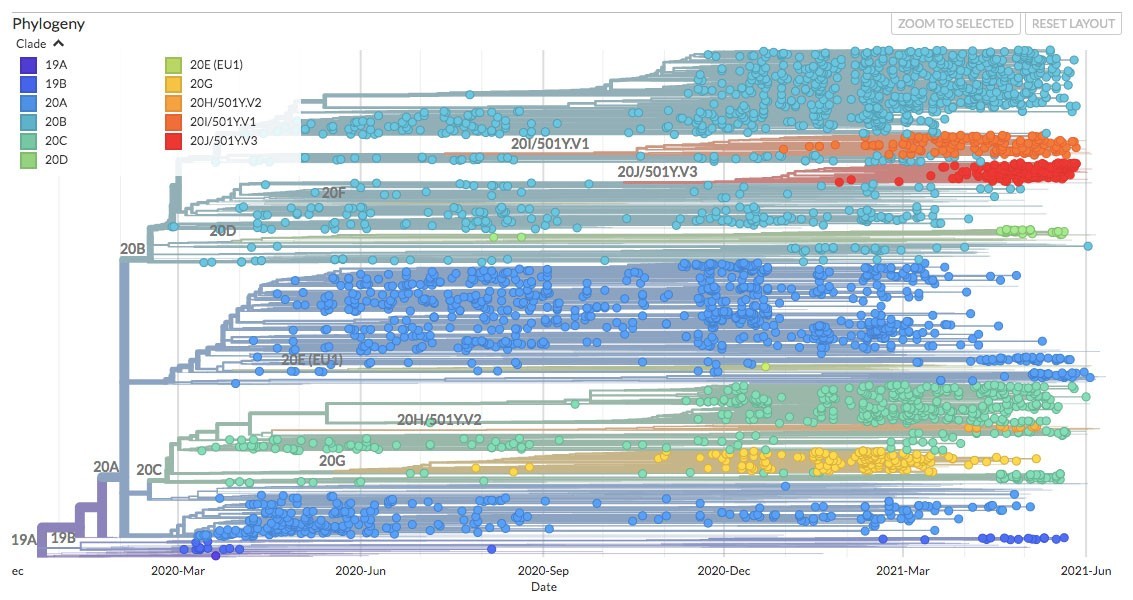
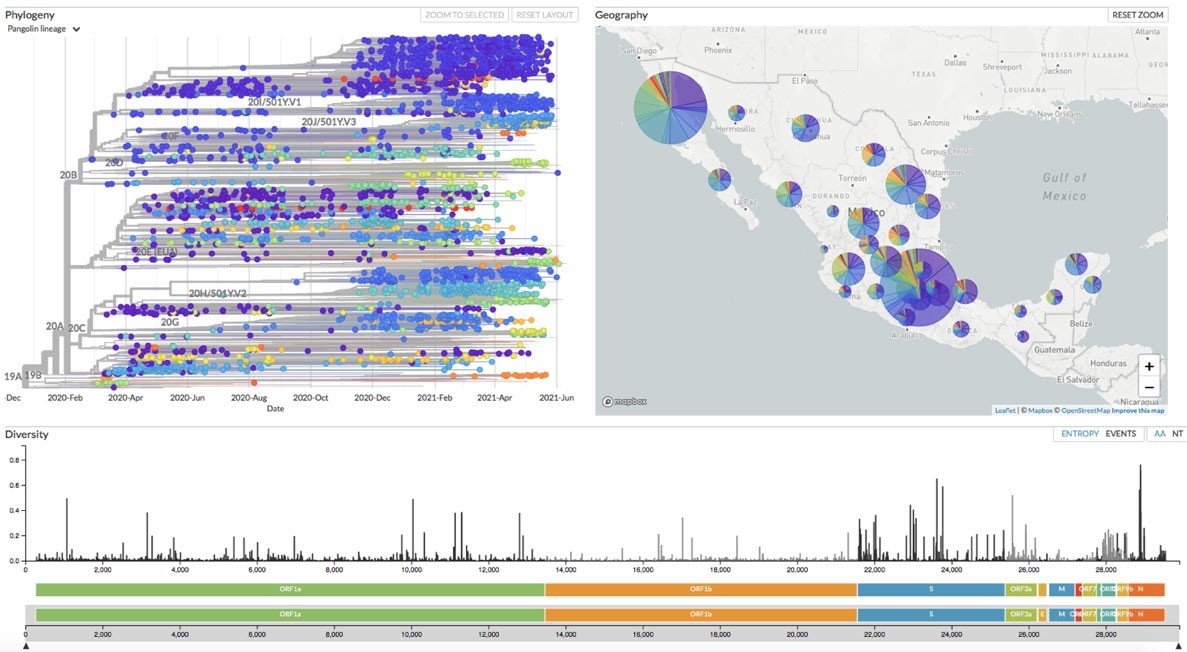
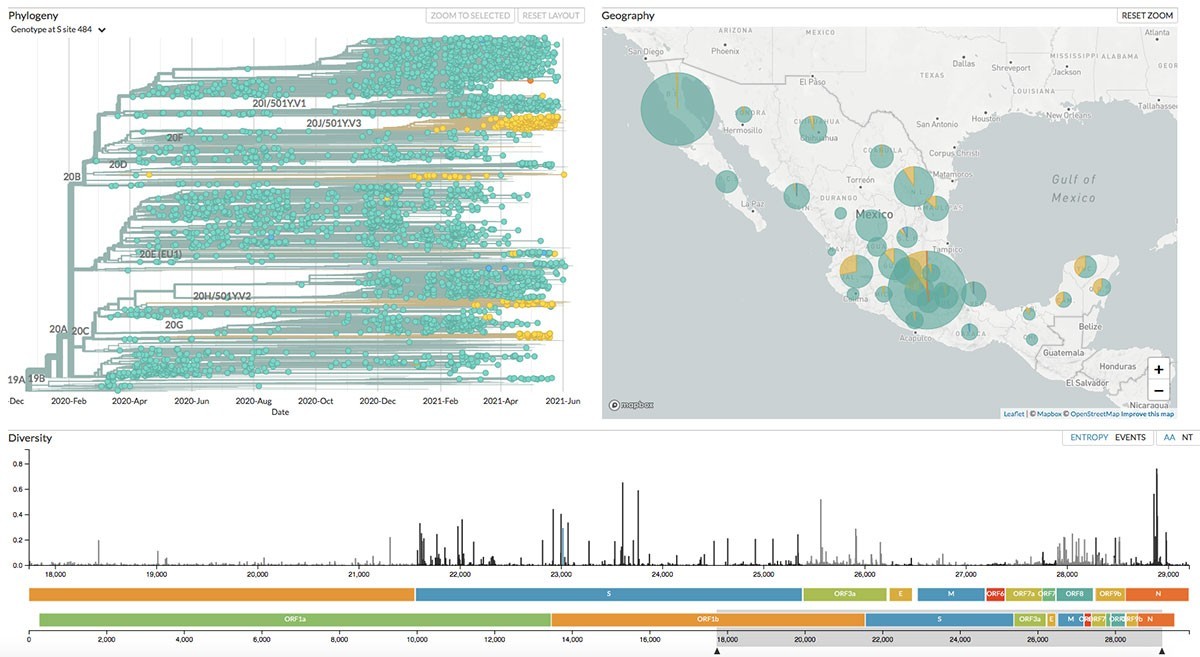
La versión de Nexstrain que implementamos en Mexstrain posee varias funciones que nos permiten estudiar la evolución del coronavirus SARS-CoV-2 en México. En primer lugar, muestra un panel con la filogenia (es decir, la historia evolutiva) del SARS-CoV-2 (Figura 2). En ella, cada punto representa un genoma en el tiempo (como se puede apreciar, el eje de las X contiene las fechas desde el inicio de la pandemia hasta la actualidad). La filogenia es una suerte de genealogía a gran escala, en donde podemos ver las relaciones de parentesco entre las distintas variantes. En ella, apreciamos los diferentes linajes del SARS-CoV-2 que se han descrito, incluyendo las variantes de preocupación, representadas en diversos colores. Las variantes de preocupación son más infecciosas y/o riesgosas para la salud humana. En resumidas cuentas, la filogenia nos permite ordenar en el tiempo el surgimiento de las variantes del coronavirus.
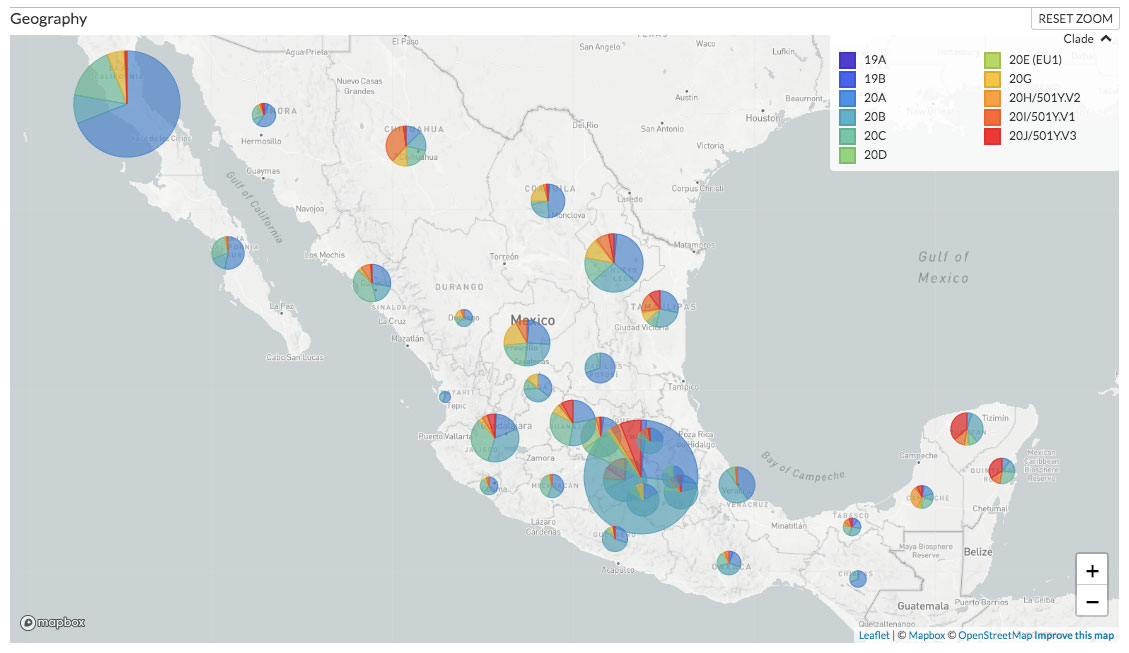
En segundo lugar, Mexstrain muestra un mapa de la República Mexicana en donde observamos la distribución de las variantes en México (Figura 3). Dependiendo del detalle con el cual se hayan registrado los lugares de muestreo, es posible ver la distribución de las variantes en el territorio nacional por estado o por ciudad.
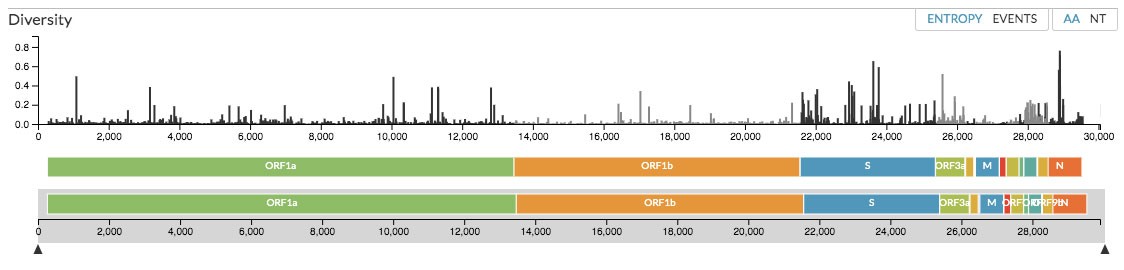
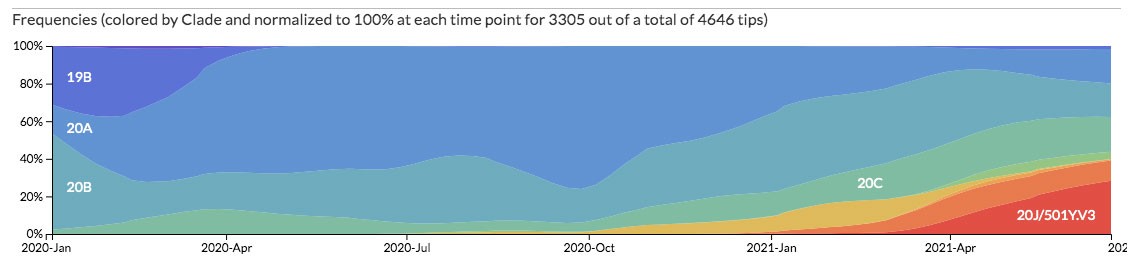
En tercer lugar, nos muestra un mapa lineal del genoma del coronavirus, desde la primera base hasta la última (Figura 4). En este mapa genómico apreciamos la localización de los genes, así como la diversidad genética asociada con ellos. Las barras verticales negras reflejan la diversidad nucleotídica asociada con cada posición en el genoma. También es posible observar la diversidad de los aminoácidos codificados. Finalmente, Nextsrain nos despliega un gráfico en donde apreciamos la frecuencia relativa de las distintas variantes a lo largo del tiempo (Figura 5).
Información cruzada
Uno de los aspectos más interesantes de la plataforma Nexstrain es que permite cruzar la información y visualizar el resultado. Por ejemplo, podemos pedirle que nos presente solo los genomas de SARS-CoV-2 que se han muestreado en México eligiendo a nuestro país en la sección Filter by Country. A continuación, le podemos pedir que nos clasifique los coronavirus que hay en México de acuerdo con el sistema de clasificación de linajes de Nextstrain. Ello lo hacemos desplegando el menú del margen izquierdo Color By y seleccionando la opción Clade. Observaremos que el número de puntos (genomas) en la filogenia disminuyó (pues señala solo a los muestreados en México) y además coloreó los linajes de acuerdo con la clasificación de Nexstrain. De igual forma, en el panel de frecuencias observamos cómo el linaje 20J/501Y.V3 (en rojo) ha incrementado su frecuencia en el país.
También podemos utilizar otro sistema de clasificación de linajes más fino si ahora elegimos Pangolin lineage en el menú Color By. El sistema de clasificación Pangolin (https://cov-lineages.org/index.html) es utilizado por científicos de todo el mundo para clasificar a las variantes del SARS-CoV-2. Veremos en el panel de frecuencias que uno de los que más ha incrementado su frecuencia en fechas recientes es el P.1. Cabe mencionar que tanto 20J/501Y.V3 como P.1 son el mismo linaje y corresponden a la variante de preocupación que se originó en Brasil y que ahora se denomina Gama por la Organización Mundial de la Salud (para saber más de las características de estos linajes virales podemos visitar la página: https://covariants.org). En cualquier caso, Nexstrain automáticamente ajusta los colores en los distintos paneles cuando cambiamos los parámetros de visualización (Figura 6).
Además, podemos buscar si alguno de los linajes que circulan en México ha adquirido determinadas mutaciones que se sabe, cambian el comportamiento del coronavirus. Por ejemplo, si en la opción Color By elegimos Genotype y en el recuadro inmediatamente inferior ingresamos la letra S (que corresponde al gen que codifica para la proteína Spike o Espiga) y además escribimos el número 484 en el siguiente recuadro, visualizaremos la variación genética asociada con el aminoácido 484 de la proteína Spike. Es sabido que la mutación E484K (en donde E representa el aminoácido ancestral y K el nuevo) le permite al coronavirus escapar con mayor facilidad de los anticuerpos fabricados por el sistema inmune. Como apreciamos en la filogenia (Figura 7), esta mutación ha aparecido por lo menos ocho veces en la evolución. Esta convergencia es una señal de evolución adaptativa. Es decir, dado que la mutación le confiere una ventaja al coronavirus, esta se selecciona recurrentemente cuando aparece en una población.
Como mencionamos más arriba, Mexstrain también utiliza la tecnología de Microreact. Una de las principales ventajas de Microreact con respecto a Nexstrain, es que nos permite visualizar muchos más genomas del SARS-CoV-2. Al momento de escribir este artículo, se han secuenciado poco más de 10,000 genomas del SARS-CoV-2 en México, los cuales están consignados en la plataforma GISAID (https://www.gisaid.org/). La plataforma Nexstrain nos permite visualizar aproximadamente 5,000 genomas, en tanto que con Microreact podemos representar la diversidad genética de las 10,000 secuencias. La versión de Microreact de Mexstrain está enfocada en mostrar la diversidad genética de las variantes de preocupación que ha declarado la Organización Mundial de la Salud.
Como vemos, las tecnologías de Microreact y Nexstrain nos permiten organizar la información genómica del SARS-CoV-2 en México. Son una herramienta invaluable para mantener una vigilancia genómica de la epidemia del coronavirus. Sin embargo, es importante tener en cuenta sus limitaciones. Tal vez la más importante es que la información que nos despliegan representa sólo una pequeña muestra de la realidad epidemiológica del país. De ahí la importancia de los esfuerzos de secuenciación realizados en todo el país. En última instancia, Mexstrain tiene el objetivo de democratizar el conocimiento de la evolución del SARS-CoV-2 en México y complementar otros esfuerzos que se realizan en el mismo sentido, tales como el efectuado por el Consorcio Mexicano de Vigilancia Epidemiológica (http://mexcov2.ibt.unam.mx:8080/COVID-TRACKER/). Además, Mexstrain es de código abierto, lo que quiere decir que cualquier investigador puede descargar los programas en la plataforma https://github.com/luisdelaye/Mexstrain y realizar sus propios análisis.
Coda
Finalmente, una pequeña reflexión. En algunas ocasiones se ha planteado una disyuntiva entre realizar ciencia aplicada o ciencia básica. En particular, se ha argumentado que, en un país como el nuestro, en dónde existen carencias de todo tipo, los científicos deberíamos enfocarnos en realizar sólo investigación aplicada. Dejando de lado el contrargumento de que es mejor hacer ciencia con ambos enfoques: básicos y aplicados. Me gustaría hacer notar que los conceptos y los métodos que nos permiten organizar en el tiempo la información genómica del SARS-CoV-2 en un árbol filogenético para entender su evolución, derivan directamente de lo que podríamos denominar ciencia básica. Sin embargo, en el contexto pandémico en el que nos encontramos, tienen una aplicación directa en la salud de la población. Es un ejemplo más de que no podemos predecir qué conocimiento nos será útil y cuál no. Citando a quien fuese mi Director de Doctorado: “La dicotomía entre ciencia básica y aplicada es falsa, debemos de preocuparnos por hacer ciencia de calidad… y si la ciencia es buena, entonces está podrá tener aplicaciones en el futuro”.
Ligas:
Mexstrain se puede consultar en: http://www.ira.cinvestav.mx/ncov.evol.mex.aspx