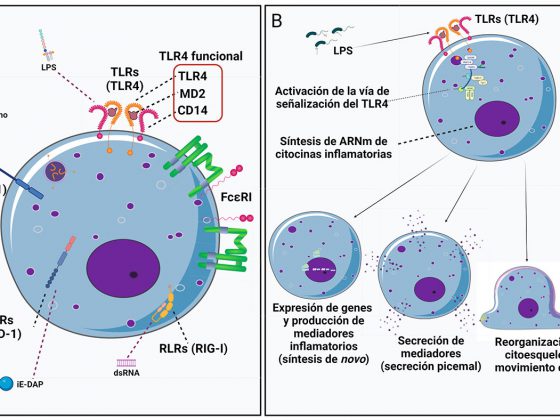
Figura 1. Mecanismo de protección pulmonar con apratastat y TMI-1. En el pulmón sano, ACE2 genera dos metabolitos activos que protegen contra la inflamación excesiva y daño tisular (izquierda). La COVID-19 favorece la activación de ADAM17, que provoca la disminución de la función de ACE2. Además, se aumenta la secreción de citocinas proinflamatorias (IL-6, IL-1β, TNF-α) culminando en una tormenta de citocinas acompañada de un reclutamiento excesivo de neutrófilos (morado) y macrófagos (amarillo) (medio). La administración de un inhibidor de ADAM17 (apratastat o TMI-1) protege contra la tormenta de citocinas atenuando el reclutamiento de neutrófilos y macrófagos, y el daño tisular (derecha).
El coronavirus del síndrome respiratorio agudo severo 2 (SARS-CoV-2) es el causante de la enfermedad por coronavirus (COVID-19), que apareció en China en diciembre de 2019. A la fecha, esta enfermedad ha causado 6.11 millones de decesos alrededor del mundo, de los cuales 322 000 ocurrieron en México (2). La enfermedad se presenta con cuadros clínicos que van de leves a moderados y en algunos casos progresan a graves. Esto es debido a que la COVID-19 induce daño pulmonar que puede culminar en SARS y falla multiorgánica que causa la muerte (3). La presencia del SAR-CoV-2 en el pulmón desencadena una rápida y coordinada respuesta defensiva del sistema inmune. Sin embargo, cuando el virus persiste y logra multiplicarse en el cuerpo, se llega a una fase grave de COVID-19, donde la respuesta inmune se descontrola y se convierte en lo que se conoce como “tormenta de citocinas”, caracterizada por una rápida y alta producción de citocinas proinflamatorias y reclutamiento excesivo de células inmunes agresivas como son los neutrófilos a los órganos, contribuyendo al daño tisular (4). Además, disminuye el número de linfocitos (linfopenia) y aumenta el número de neutrófilos (neutrofilia) en la sangre, un fenómeno característico de la COVID-19.
El SARS-CoV-2 entra en la célula huésped a través de la unión entre su proteína espiga (Spike; S) con el receptor de la enzima convertidora de angiotensina 2 (ACE2) del huésped, receptor presente en células del pulmón, intestino delgado y de los vasos sanguíneos (7). La ACE2 es una enzima que produce hormonas que protegen contra la inflamación excesiva y el daño tisular (Figura 1) (9). Durante la COVID-19 la concentración de la ACE2 disminuye por enzimas que la degradan (10), incluyendo una desintegrasa y metaloproteasa 17 (ADAM17) (11). Así, el virus reduce los efectos protectores de ACE2 en diferentes órganos incluyendo el pulmón. Además, ADAM17 también produce la forma activa de la citocina proinflamatoria, factor de necrosis tumoral-alfa (TNFα), contribuyendo a la tormenta de citocinas y al excesivo reclutamiento de neutrófilos (13). Derivado de esta información, consideramos importante estudiar el papel de ADAM17 durante la COVID-19 y analizar si ADAM17 pudiera ser propuesto como un blanco terapéutico. Actualmente las terapias para los pacientes con COVID-19 se basan principalmente en tratamientos antivirales y antiinflamatorios, pero ningún régimen está, en particular, dirigido a la prevención y recuperación de los daños en órganos como el pulmón.
Para investigar los efectos de la inhibición de ADAM-17 en la inflamación pulmonar vinculada con la COVID-19, establecimos un modelo preclínico en ratones que simula el daño pulmonar agudo causado por esta enfermedad, el cual consta de la administración intratraqueal de una porción de la proteína S del SARS-CoV-2 (dominio RBD de la proteína Spike; proporcionado por el Dr. Edgar Morelos del Departamento de Bioquímica, Cinvestav-IPN) en combinación con el poly(I:C), una molécula asociada con infecciones virales. Usando diferentes métodos como histología y citometría de flujo encontramos, que la combinación de RBD/poly(I:C) aumentó la producción de citocinas proinflamatorias y el reclutamiento de células inmunes como neutrófilos y macrófagos al pulmón, causando daño pulmonar considerable (13). Tratando a los ratones enfermos con los inhibidores de ADAM-17, apratastat y TMI-1, descubrimos que la presencia de neutrófilos y macrófagos disminuyó en los pulmones, en paralelo con la inhibición del aumento de neutrófilos circulantes en la sangre periférica. Ello se debe a la disminución en la producción de citocinas proinflamatorias y al aumento de citocinas antiinflamatorias en los ratones enfermos tratados con apratastat y TMI-1. Se destaca la observación de menos daño en los pulmones de los ratones enfermos que recibieron apratastat y TMI-1; y ningún efecto adverso en ratones sanos tratados con estos fármacos (13).
Resumiendo, hemos proporcionado evidencia experimental de que la inhibición de ADAM-17 por apratastat y TMI-1 reduce el daño pulmonar, la tormenta de citocinas y el reclutamiento de neutrófilos a los pulmones en un modelo murino de COVID-19 (Figura 1). Proponemos el uso de apratastat y TMI-1 en ensayos clínicos para evaluar su eficacia en pacientes con la COVID-19 como tratamiento para prevenir que una infección con SARS-CoV-2 progrese a su forma grave.
Bibliografía
1.- WHO. Clinical management of severe acute respiratory infection when Novel coronavirus (nCoV) infection is suspected: interim guidance. Jan 11, 2020. https://www.who.int/internalpublications-detail/clinical-management-of-severe-acute-respiratoryinfection-when-novel-coronavirus-(ncov)-infection-is-suspected (accessed Jan 20, 2020).
2.- Johns Hopkins University Center for Systems Science and Engineering (JHU CSSE) https://www.arcgis.com/apps/dashboards/bda7594740fd40299423467b48e9ecf6
3.- Chen N, Zhou M, Dong X, et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. The Lancet 2020 Volume 395: 507-513
4.- Rafael Miranda Pedroso. Tormenta de citoquinas en la infección por SARS-CoV-2 (COVID-19). Revista Cubana de Medicina Intensiva y Emergencias. 2021;20(3):e830
5.- Vadillo E, Taniguchi-Ponciano K, Lopez-Macias et al. A Shift Towards an Immature Myeloid Profile in Peripheral Blood of Critically Ill COVID-19 Patients. Arch Med Res. 2021:52(3):311-323.
6.- Matthay MA, Wick KD. Corticosteroids, COVID-19 pneumonia, and acute respiratory distress síndrome. J. Clin Invest. 2020;130:6218-6221.
7.- Hoffmann M, Kleine-Weber H, Schroeder S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271-280e278.
8.- Angiotensin converting enzyme 2 and its emerging role in the regulation of the renin angiotensin system. María José Solera, Josep Lloverasa, Daniel Batlleb Medicina Clinica. 2008;131: 230-236.
9.- GheblawiM, Wang K, Viveiros A,etal.Angiotensin-converting enzyme2:sARS-CoV-2 receptor and regulator of the renin-angiotensinsys-tem: celebrating the 20th anniversary of the discovery of ACE2. Circ Res .2020; 126:1456-1474.
10.-Kuba K, Imai Y, Rao S, et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung in jury. Nat. Med. 2005;11:875-879.
11.-Zunke F, Rose-John S. The shedding protease ADAM17: physiology and pathophysiology. Biochim Biophys Acta Mol Cell Res.2017;1864:2059-2070.
12.- Cui SN, TanHY, Fan GC. Immunopathological roles of neutrophil sin virus infection and COVID-19. Shock.2021.
13.- Lartey NL, Valle-Reyes S, Vargas-Robles H et al. ADAM17/MMP inhibition prevents neutrophilia and lung injury in a mouse model of COVID-19. J Leuk. Biol. 2021: 26. doi: 10.1002/JLB.3COVA0421-195RR