La investigación química no ocurre sólo dentro de un laboratorio. En nuestros días, la química sigue siendo una ciencia ampliamente experimental en donde se combinan sustancias de distinta naturaleza y se modifican las condiciones en que las reacciones químicas se realizan con fines específicos. No obstante, en nuestra era, existe otra área menos conocida de la química, pero igualmente importante. Ésta se lleva en primera instancia en una computadora para modelar, visualizar y cuantificar comportamientos complejos mediante simulaciones establecidas sobre una teoría mínima, para comprender primeramente y después modificar aspectos de un diseño experimental. Esta área es conocida como Química Computacional y ha estado detrás de diversos logros en los siglos XX y XXI. Así, trabajos en este ámbito han hecho acreedores al premio Nobel a Walter Kohn y John A. Pople en 1998, y a Martin Karplus, Michael Levitt y Arieh Warshel en 2013.
Durante la pandemia de COVID-19, la Química Computacional mostró su valía permitiendo entender mejor el mecanismo de acción química del virus y así diseñar vacunas más efectivas. Esto hubiera sido difícil de lograr por prueba y error en el laboratorio, modificando los diferentes parámetros que intervienen, sin una simulación computacional de las propiedades en la acción bioquímica y los efectos finales deseados. Así, las simulaciones computacionales, sustentando el enfoque operativo de la disciplina, se emplean ampliamente en la comprensión de cómo surgen las propiedades de los materiales a nivel molecular, explotarlas y potenciarlas para que resulten mejoradas. Ejemplos de ello son la capacidad electroquímica para lograr baterías más ligeras y duraderas, o el diseño de materiales fotovoltaicos con mejor eficiencia en la conversión de energía solar en eléctrica. Con anterioridad, el enfoque puramente experimental y tradicional solía ser empírico y por eso menos eficaz.
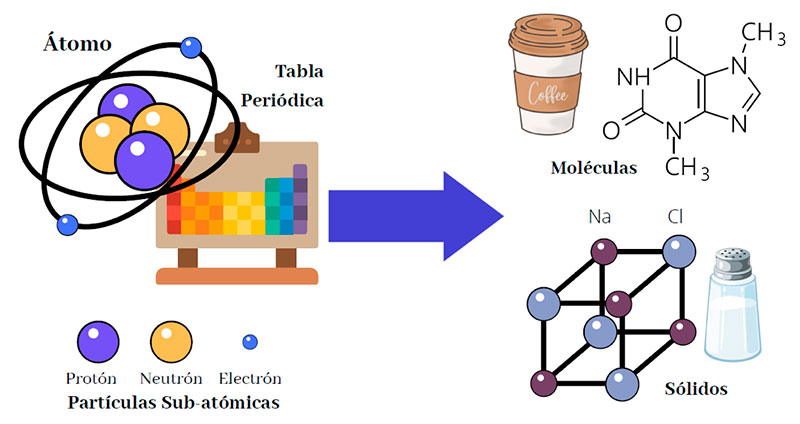
Para comprender más sobre cómo opera la Química Computacional y sus usos en investigación, es importante conocer algunos aspectos esenciales de la química. Gran parte de la materia que manipulamos en nuestra vida diaria la constituyen átomos conformados por partículas subatómicas como electrones, protones y neutrones en un primer nivel. Entre ellas median interacciones primordialmente eléctricas o bien otro tipo de fuerza entre protones y neutrones, como la denominada fuerza nuclear fuerte, responsable de la estabilidad de dichas estructuras, formando así al átomo y su núcleo como sistemas estables dentro de un rango de condiciones habituales en nuestra vida cotidiana. Al combinarse, los átomos originan estructuras superiores como las moléculas, los sólidos cristalinos y gran parte de la materia que conforma nuestra experiencia diaria, como se muestra en la Figura 1. Todas estas estructuras, su forma, la manera en que geométricamente se componen, disponen y combinan, pueden depender de las reglas que operan entre las interacciones mencionadas. Por ello, para comprender su variabilidad, hoy en día las exploramos y estudiamos mediante simulaciones, para luego experimentar con ellas a partir de una aproximación previa difícil de cuantificar a partir solo de sus propiedades macroscópicas, como lo hacía la química tradicional. Sin embargo, en algunos casos estas interacciones en los ámbitos molecular o atómico no son suficientes, pues su estudio se limita a aproximaciones físicas dictadas sólo por teorías e interacciones clásicas como el electromagnetismo u otros modelos empíricos observados como supuestas buenas aproximaciones, sin involucrar más profundamente a las leyes de la física.
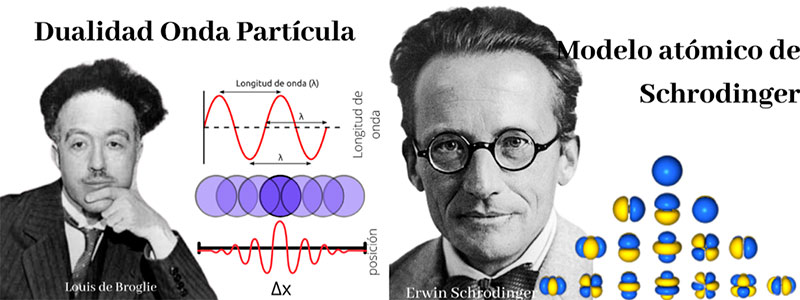
Durante el siglo pasado, la física y la ciencia en su conjunto atravesaron por una revolución que cambió la manera de entender al universo, y que dio lugar a discrepancias en nuestras predicciones a diversas escalas: la teoría cuántica. Ésta postula que la materia, dependiendo de las circunstancias, puede comportarse como onda o partícula. El físico Louis de Broglie estableció en ese contexto una base teórica para el electrón, haciéndose por ello acreedor al premio Nobel en 1929. Con esta descripción más precisa del mundo, las partículas, y en general los sistemas cuánticos, se rigen por la ecuación de Schrödinger. Estos dos hechos se muestran en la Figura 2. Gracias a Schrödinger se estableció la base teórica para el hecho empíricamente ya conocido de que los electrones en los átomos ocupan niveles de energía definidos, conocidos como orbitales atómicos. Esta teoría modificó la imagen clásica de los electrones moviéndose alrededor del núcleo atómico como un sistema planetario. Cuando los átomos se combinan para formar moléculas, los orbitales atómicos conforman nuevas estructuras y dan origen a orbitales moleculares. Estas diferencias en relación con las aproximaciones clásicas de interacción entre moléculas modificaron la visión de la química y de cómo la física opera dentro de las reacciones químicas y configuraciones geométricas de las moléculas. Como en los átomos, en las moléculas estos orbitales rigen la reactividad química, sus propiedades magnéticas, los procesos ópticos de absorción y emisión de luz, hasta su propia conformación. Así, muchas de las propiedades específicas de la materia, debieron ser revisitadas para comprender mejor o descubrir fenómenos como la fluorescencia, la fosforescencia y la fotocromaticidad.
Aunque la teoría cuántica nos ha provisto de las ecuaciones necesarias para describir mucho mejor y más ampliamente un sinfín de fenómenos químicos, cuando las aplicamos a sistemas tan complejos como moléculas orgánicas e incluso inorgánicas, resulta imposible resolverlas de manera exacta para predecir aspectos que van desde su configuración geométrica, su comportamiento dinámico, su reactividad química y otras propiedades fisicoquímicas. Sin embargo, los físicos y químicos no se han dado por vencidos ante esta limitación y han recurrido a aproximaciones matemáticas, algoritmos computacionales sofisticados y el uso de supercomputadoras muy rápidas para analizar el comportamiento de estos sistemas mediante simulaciones numéricas, dando origen a la Química Computacional como un área de desarrollo e investigación. La forma de trabajar es ilustrada en la Figura 3: un ciclo recursivo que permite establecer premisas, simularlas, obtener comportamientos esperados, experimentarlos, modificar las premisas y repetir el proceso de cálculo. Hoy en día, la Química Computacional está al alcance de muchas más personas con cierta preparación en química, pues permite utilizar y explotar diversas herramientas computacionales en línea que implementan algoritmos de simulación avanzados, sin preocuparse en extremo por las matemáticas, la física y la computación sobre las que éstas se sustentan. Por ejemplo, podemos explorar diversas propiedades que estudian los químicos teóricos utilizando software de uso libre, como ORCA, una aplicación en línea que aplica propiedades cuánticas a la simulación química y permite el análisis computacional de diversas estructuras a partir de principios mínimos básicos, denominados ab initio. Así, es posible calcular numéricamente la curva de energía de una molécula de etano sujeta a deformaciones por la interacción con otras estructuras químicas, e incluso analizar cómo se modifican las propiedades de absorción de luz de una molécula cromófora en la región UV o visible en función de sus parámetros estructurales.
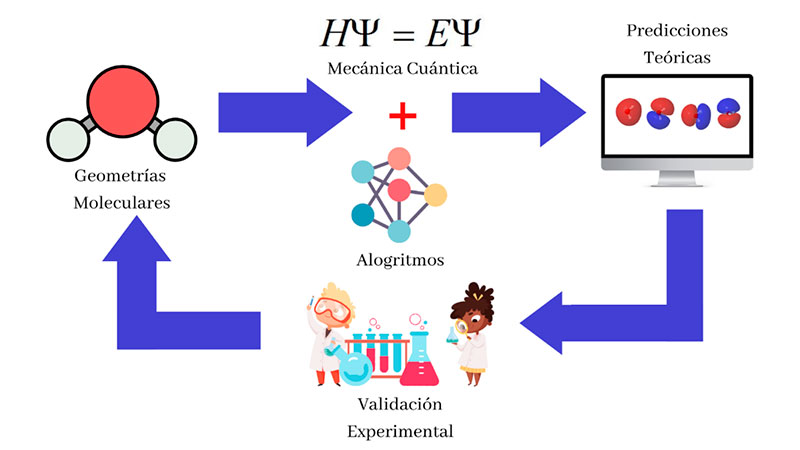
A pesar de que la Química Computacional ha probado su utilidad dentro de la investigación sobre propiedades químicas de la materia, existen aún retos que limitan este enfoque en forma extendida. El primero de ellos es el impuesto por el tiempo de cómputo necesario para obtener las predicciones, el cual crece exponencialmente con el número de átomos de una configuración o molécula a estudiar. En muchos casos resulta prácticamente imposible el uso de metodologías de la Química Computacional en áreas como la bioquímica, donde las proteínas poseen entre cientos y miles de aminoácidos, lo cual, por lo expresado antes, implican tiempos de procesamiento muy grandes, como meses o años inclusive. Por otro lado, existe un problema fundamental, la correlación electrónica, una propiedad ampliamente conocida como entrelazamiento cuántico, por la que los componentes individuales de un sistema adquieren propiedades de grupo. Esta propiedad determina en muchas estructuras el transporte de energía en aquellos complejos que intervienen en la fotosíntesis, por lo que la inclusión de su descripción cuántica resulta crucial. Las computadoras tradicionales no pueden modelar naturalmente estas interacciones que surgen entre los electrones, aumentando con ello la complejidad computacional y el tiempo de procesamiento necesarios.
De tal suerte, así como se han buscado problemas como los anteriores, se han buscado nuevas formas para reproducir el comportamiento químico de muchos compuestos moleculares que implique las predicciones y efectos inducidos por la mecánica cuántica. Entre estos, la simulación empleando sistemas cuánticos análogos ofrece una nueva aproximación al cálculo de las propiedades químicas de la materia involucrando la complejidad cuántica. A este enfoque se le conoce como procesamiento cuántico: la utilización de sistemas cuánticos cuyo comportamiento físico es bien conocido, para emular el comportamiento de otros sistemas más complejos. Ello parte de la premisa de que existen sistemas que entendemos a detalle y que es posible manipular. Partiendo de estos sistemas modelo, es posible reproducir el comportamiento de sistemas más complicados en un lenguaje más natural que ya incluye las propiedades cuánticas. Esta idea fue inicialmente sugerida por el físico Richard Feynmann, ganador del premio Nobel en 1965 por sus desarrollos en electrodinámica cuántica. Pasaría mucho tiempo para que esta idea no fuera considerada como ciencia ficción en vez de una posibilidad real. Hoy en día nuestra capacidad tecnológica en el ámbito cuántico ha aumentado notablemente y comenzamos a hacer posible llevar estas ideas a la práctica. Algunos de los sistemas cuánticos empleados para este propósito incluyen los átomos ultra fríos, las trampas de iones y los láseres, utilizados para establecer arquitecturas de procesadores cuánticos en el sentido mencionado.
El procesamiento cuántico ha surgido como un modelo de computación viable que explota estos sistemas y sus propiedades, como el entrelazamiento cuántico y la superposición de estados. En ésta, a diferencia de los bits de las computadoras personales, los sistemas pueden encontrarse en un estado indefinido o de superposición de dos o más estados físicos, que identificados con el “0” y el “1”, permiten establecer bits cuánticos o qubits (Figura 4) y una analogía potencialmente más poderosa que sus contrapartes tradicionales. Algunos sistemas cuánticos que exhiben este comportamiento son los fotones a través de la polarización de la luz o el espín de un electrón, átomo o molécula (Figura 5).
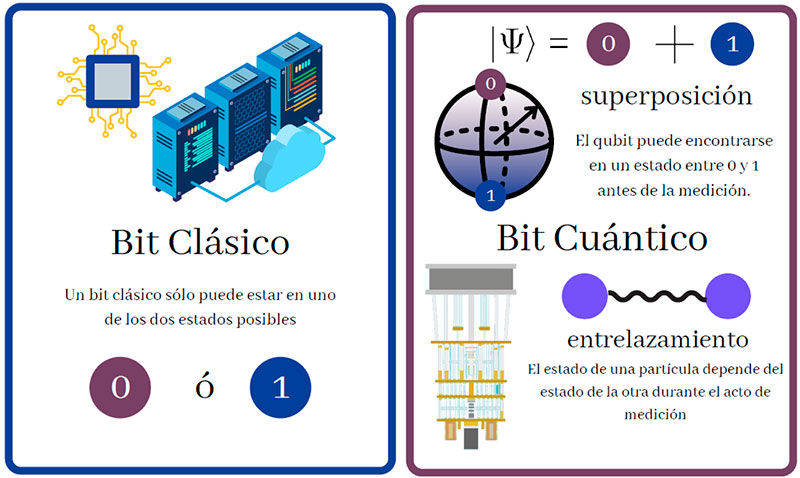
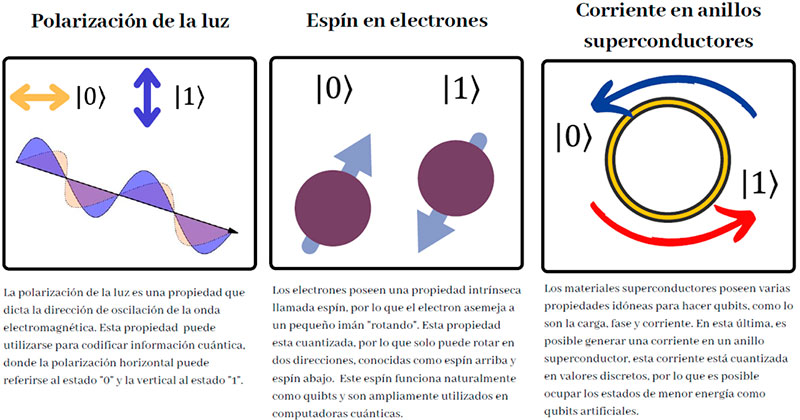
Haciendo interactuar estos sistemas (por ejemplo, mediante campos electromagnéticos o ciertos tipos de materiales ópticos), podemos originar cambios controlados, operaciones sobre la información guardada en ellos que permiten transformarla. Estas interacciones se diseñan para generar operaciones específicas que emulan a aquellas del cómputo tradicional; son conocidas como compuertas cuánticas, lo que permite sintetizar circuitos de procesamiento cuántico y con ello, algoritmos, sin la necesidad de conocer los detalles de cómo se llevan a cabo dichas operaciones y concentrándose en su efecto sobre la información almacenada.
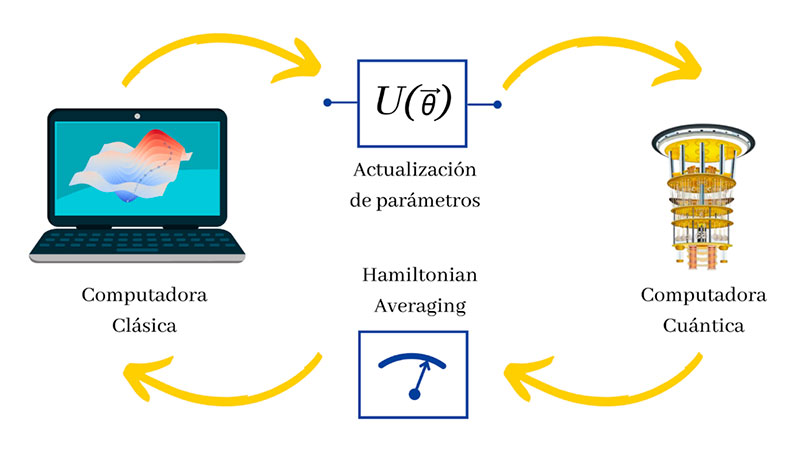
Las computadoras cuánticas no se consideran remplazo de las computadoras tradicionales de hoy en día, sino como sistemas que en conjunto ayudan a resolver cuestiones complejas sobre el mundo que nos rodea. Entre los algoritmos cuánticos propuestos para la simulación de sistemas químicos destaca el algoritmo Cuántico Variacional o VQE por sus siglas en inglés (Variational Quantum Eigensolver Algorithm). Éste se puede instrumentar en el hardware de naturaleza cuántica disponible (Station Q o IBM Q, por ejemplo). Es un algoritmo híbrido, ya que emplea tanto una computadora cuántica para ciertos cálculos, como una tradicional para otros.
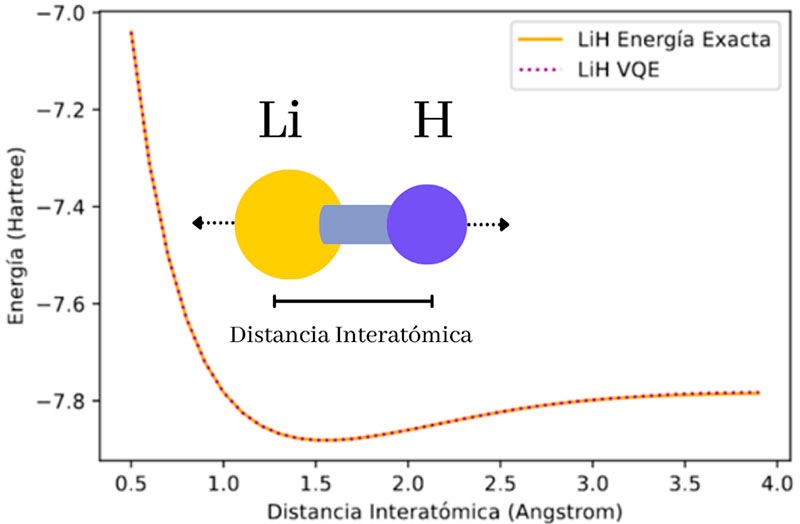
En este algoritmo, el circuito cuántico es un conjunto de compuertas controladas, las cuales pueden cambiar los estados de algunos qubits en función de otros, así como generar entrelazamiento cuántico entre pares de qubits, lo que permite calcular la energía de una molécula considerando algunas de sus propiedades cuánticas. Para ello, el problema se codifica a través de qubits mediante un proceso conocido como Hamiltonian averaging. En tanto, un optimizador tradicional se encarga de encontrar una serie de nuevos parámetros que actualizan la información en el propio circuito cuántico, optimizando la energía calculada para una siguiente evaluación. El algoritmo se repite hasta encontrar un valor de energía convergente, el cual será el de la molécula que se estudie (Figura 6). En este enfoque, algunas herramientas de libre uso como Qiskit o Pennylane (ver las referencias de estos recursos), poseen librerías que permiten simular circuitos cuánticos como el VQE. Por ejemplo, en Qiskit podemos calcular desde nuestra propia computadora la energía del estado de energía mínimo de moléculas diatómicas, como la molécula de LiH (Hidruro de Litio), así como obtener las superficies de energía que se forman al distorsionar el enlace de esta molécula (Figura 7).
Con el procesamiento cuántico, la Química Computacional se aproxima a una nueva revolución química donde podremos diseñar en un tiempo récord y con una diversidad que el cómputo tradicional no ha permitido, nuevos y más sofisticados materiales para la salud, la seguridad y la vida cotidiana en general. Hoy más que nunca podemos ser parte de la revolución cuántica desde la investigación química, bioquímica y biológica.
Referencias
Gribbin, J. 1984. En busca del gato de Schrödinger. Biblioteca Científica Salvat: México, México.
Vedral, V. 2011. Descodificando la realidad: el universo como información cuántica. Biblioteca Buridan: Madrid, España.
Cuevas, G. 2005. Química computacional. Revista Ciencia. Academia Mexicana de Ciencias. Consultar en: https://www.revistaciencia.amc.edu.mx/images/revista/56_2/quimica_comutacional.pdf
Kaye, P., Laflamme, R. and Mosca, M. 2011. An introduction to quantum computing. Oxford University Press: New York, USA.
Anaya, A. and Delgado, F. 2019. Benchmarking of Adiabatic Quantum Computation models to predict the structure of proteins. Journal of Physics: Conference Series, 1391, 012016.
Anaya, A. and Delgado, F. 2020. Using the Energy Distribution as a Benchmarking Among Models for Adiabatic Quantum Processing in the Protein Folding Problem. Journal of Physics: Conference Series, 1540, 012032.
Anaya, A. and Delgado, F. 2022. Simulating molecules using the VQE algorithm on Qiskit. ArXiv:2201.04216v1 [quant-ph]
FAccTs, 2022. ORCA tutorials – Compatible with ORCA 5.0! Consultar en: https://www.orcasoftware.de/tutorials_orca/
Qiskit Development Team, 2022. Simulating Molecules using VQE. Consultar en: https://qiskit.org/textbook/ch-applications/vqe-molecules.html
Xanadu, 2019. Quantum Chemistry. Consultar en: https://pennylane.ai/qml/demos_quantum-chemistry.html