

Las proteinopatías son un grupo de enfermedades que se originan por la presencia de proteínas con mutaciones en las células, lo que altera su organización y lleva a su acumulación anormal en forma de agregados proteicos, derivando en un mal funcionamiento celular, y en los casos más graves, genera enfermedades neurodegenerativas.1 Además de los defectos en la síntesis proteica, estas patologías también se originan por errores en el plegamiento, el mantenimiento de la conformación y los mecanismos de la degradación de proteínas.2
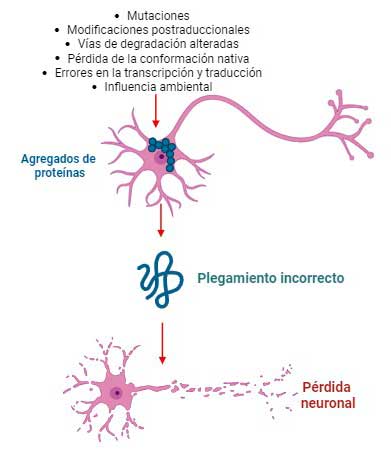
La esclerosis lateral amiotrófica (ELA) o también llamada enfermedad de la neurona motora3 es una proteinopatía y una enfermedad neurodegenerativa del sistema nervioso central que es mortal.4 La ELA se caracteriza por la muerte de las neuronas motoras del cerebro, del tronco del encéfalo y de la médula espinal,5 lo que conlleva a que los músculos desarrollen poca o nula fuerza,6 y finalmente conduce a la muerte de los pacientes por insuficiencia respiratoria, en un plazo de 3 a 5 años después del inicio de los síntomas7.
Desafortunadamente, el tratamiento de la enfermedad aún dista mucho de ser efectivo, ya que el riluzol, uno de los fármacos aprobados para tratar la ELA, además de su alto costo, sólo incrementa 3 meses en promedio la supervivencia de los pacientes.8
La incidencia global estandarizada de la ELA es de 4.42 por 100.000,9 con un aumento progresivo asociado a la edad, alcanzando su máximo entre los 60 y 79 años.4 La edad media de aparición es a los 62 años.10 Además, existe una disparidad por sexo, con un predominio masculino y una tasa entre hombres y mujeres de 1.35:1.4
Los pacientes con ELA poseen varios genes con mutaciones, los cuales dan lugar a proteínas alteradas que se “pegan” entre sí, llegando a formar agregados que contribuyen a la fisiopatología de esta enfermedad.11
A continuación, se enlistan algunas de las proteínas y genes con mutaciones que se presentan en pacientes con ELA.
Proteína 43 de unión a DNA de respuesta transactiva (TDP-43/TARDBP)
La proteína 43 de unión a DNA de respuesta transactiva (TDP-43) es una proteína cuya función es unirse al ADN y ARN, en donde se involucra con varios procesos celulares, tales como la transcripción en la que se transfiere información genética del ADN a una copia de ARN mensajero (ARNm),12 su estabilidad y la traducción de su información para sintetizar proteínas. TDP-43 se expresa en neuronas y en células gliales de manera fisiológica, y se encuentra principalmente en el núcleo de la célula.13
En la ELA se incrementa el paso de la TDP-43 del núcleo al citoplasma neuronal,10 en donde forma agregados en células de la corteza cerebral y en la médula espinal, lo que hace que se acumule en exceso en tales células.11 Aproximadamente, el 97% de los pacientes con ELA presentan proteinopatía asociada a TDP-43.3
Proteína Superóxido dismutasa 1 (SOD1)
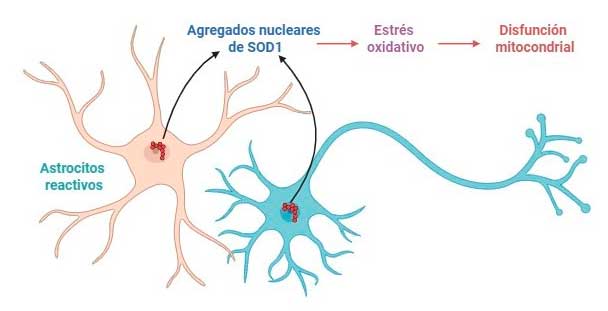
La proteína superóxido dismutasa 1 (SOD1) es esencial para proteger a las células del daño oxidativo, debido a que elimina varios radicales libres que lo generan.14 Las proteínas SOD1 mutantes presentan una ganancia en su función, por lo que tienden a ensamblarse, y sus agregados se acumulan en las neuronas motoras. Hasta la fecha, se han identificado más de 200 mutaciones distintas asociadas a alteraciones en esta proteína. Además de las mutaciones, otros factores como la oxidación y las modificaciones postraduccionales (MPTs) también promueven la formación de agregados.15 Esta acumulación causa una respuesta de estrés oxidativo debido a un desequilibrio en la generación y eliminación de radicales libres.12
Los agregados de SOD1 están presentes en el núcleo de las neuronas motoras, así como en el núcleo de astrocitos, microglía y oligodendrocitos de la médula espinal en pacientes con ELA.15
Cabe destacar que el modelo de ratón transgénico SOD1 G93A, ampliamente utilizado en la investigación preclínica de la ELA, expresa una variante humana de SOD1 con una mutación puntual (glicina a alanina en la posición 93). Este modelo simula los hallazgos histopatológicos y clínicos, incluyendo un inicio temprano y una progresión veloz de los síntomas.14
Proteína Fusionada en sarcoma (FUS)
La proteína fusionada en sarcoma (FUS) es una proteína con ubicación nuclear que se une al ARN y ADN, y su expresión se encuentra asociada con las formas más agresivas y de aparición temprana de la ELA. Esta proteína tiene la función de reparar los daños del ADN y participa en el transporte, la estabilidad y la traducción de ARNm, entre otros.16
Las mutaciones en FUS causan un transporte anormal de la proteína al citoplasma.12 La proteína mutante puede formar agregados tanto en el núcleo como en el citoplasma de las neuronas de la médula espinal y del cerebro de los pacientes con ELA. Estas inclusiones inducen la formación de gránulos de estrés en el citoplasma que impiden el procesamiento de los ARN.13
Gen C9ORF72
El gen C9ORF72 es la causa genética más común en pacientes con ELA.10 Contiene un segmento con una secuencia de nucleótidos que incluyen cuatro guaninas y dos citosinas (GGGGCC), que se presenta de 2 a 5 veces en personas sanas.12 Sin embargo, tal secuencia puede alterarse y presentarse miles de veces, provocando la ELA.11 Estas repeticiones múltiples se relacionan con una menor supervivencia y con una reducción en la edad de aparición de la enfermedad en los hombres con respecto a las mujeres.4
Funcionalmente, la proteína C9ORF72 interactúa con proteínas que procesan al ARN. De este modo, la proteína mutada genera un gran secuestro de proteínas de unión a ARN, así como la formación de estructuras hibridas de ADN-ARN que aumentan la susceptibilidad del daño al ADN y la inestabilidad del genoma.3
Proteína alfa-sinucleína (α-sinucleína)
La alfa-sinucleína (α-sinucleína) es una proteína involucrada en la función sináptica, que se encuentra de manera normal en el Sistema Nervioso Central. En condiciones patológicas, los agregados de esta proteína, denominados cuerpos de Lewy, son característicos de la enfermedad de Parkinson y de la demencia con cuerpos de Lewy (DCL).17
Sin embargo, se ha demostrado que los agregados de la α-sinucleína incrementan la agregación de SOD1, lo que sugiere un fenómeno de agregación cruzada en el que esta proteína es un desencadenante de la formación de agregados de SOD1.18 Además, estos agregados se han observado en el tronco encefálico y en el asta anterior de la médula espinal de pacientes con ELA. Por el contrario, el mesencéfalo de estos mismos pacientes, que es la región típicamente afectada en la enfermedad de Parkinson, no presenta dichos agregados.17
Recientemente se ha reportado que mutaciones puntuales en la proteína afectan su ensamblaje mediante el reemplazo de aminoácidos, lo que acelera la agregación, reduce la solubilidad de los monómeros y aumenta la secreción de α-sinucleína.19
Más allá de las mutaciones, las MPTs también pueden influir en la propagación y amplificación de la proteína patológica. Estas modificaciones consisten en la adición de grupos funcionales a los residuos de aminoácidos, lo que altera la función, ubicación, estructura, solubilidad e interacciones con células y otras proteínas. En el caso de la α-sinucleína, se han identificado diversas MPTs, entre las que se incluyen fosforilaciones, acetilaciones, glicaciones y truncamiento.20
Debido a lo anterior, la expresión de α-sinucleína se relaciona con la degeneración de las neuronas motoras. Sin embargo, aún se desconoce si sus agregados son una consecuencia de la ELA, o bien si intensifican la enfermedad al agravar otros procesos, tales como el estrés oxidativo o la disfunción mitocondrial.17
El entendimiento de todas estas proteínas y genes fundamentales establece un fundamento sólido para la creación de tratamientos específicos y personalizados. La investigación contínua en esta área abre la posibilidad de desarrollar intervenciones más efectivas para abordar las complicaciones moleculares de la esclerosis lateral amiotrófica.
Referencias
- Bayer T. A. (2015). Proteinopathies, a core concept for understanding and ultimately treating degenerative disorders?. European neuropsychopharmacology : the journal of the European College of Neuropsychopharmacology, 25(5), 713–724. https://doi.org/10.1016/j.euroneuro.2013.03.007
- Klaips, C. L., Jayaraj, G. G., & Hartl, F. U. (2018). Pathways of cellular proteostasis in aging and disease. The Journal of cell biology, 217(1), 51–63. https://doi.org/10.1083/jcb.201709072
- Hardiman, O., Al-Chalabi, A., Chio, A., Corr, E. M., Logroscino, G., Robberecht, W., Shaw, P. J., Simmons, Z., & van den Berg, L. H. (2017). Amyotrophic lateral sclerosis. Nature reviews. Disease primers, 3, 17071. https://doi.org/10.1038/nrdp.2017.71
- Feldman, E. L., Goutman, S. A., Petri, S., Mazzini, L., Savelieff, M. G., Shaw, P. J., & Sobue, G. (2022). Amyotrophic lateral sclerosis. Lancet (London, England), 400(10360), 1363–1380. https://doi.org/10.1016/S0140-6736(22)01272-7
- Goutman, S. A., Hardiman, O., Al-Chalabi, A., Chió, A., Savelieff, M. G., Kiernan, M. C., & Feldman, E. L. (2022). Emerging insights into the complex genetics and pathophysiology of amyotrophic lateral sclerosis. The Lancet. Neurology, 21(5), 465–479. https://doi.org/10.1016/S1474-4422(21)00414-2
- Grad, L. I., Rouleau, G. A., Ravits, J., & Cashman, N. R. (2017). Clinical Spectrum of Amyotrophic Lateral Sclerosis (ALS). Cold Spring Harbor perspectives in medicine, 7(8), a024117. https://doi.org/10.1101/cshperspect.a024117
- Chia, R., Chiò, A., & Traynor, B. J. (2018). Novel genes associated with amyotrophic lateral sclerosis: diagnostic and clinical implications. The Lancet. Neurology, 17(1), 94–102. https://doi.org/10.1016/S1474-4422(17)30401-5
- Mead, R. J., Shan, N., Reiser, H. J., Marshall, F., & Shaw, P. J. (2023). Amyotrophic lateral sclerosis: a neurodegenerative disorder poised for successful therapeutic translation. Nature reviews. Drug discovery, 22(3), 185–212. https://doi.org/10.1038/s41573-022-00612-2
- Xu, L., Liu, T., Liu, L., Yao, X., Chen, L., Fan, D., Zhan, S., & Wang, S. (2020). Global variation in prevalence and incidence of amyotrophic lateral sclerosis: a systematic review and meta-analysis. Journal of neurology, 267(4), 944–953. https://doi.org/10.1007/s00415-019-09652-y
- Hulisz D. (2018). Amyotrophic lateral sclerosis: disease state overview. The American journal of managed care, 24(15 Suppl), S320–S326.
- Roberts, B., Theunissen, F., Mastaglia, F. L., Akkari, P. A., & Flynn, L. L. (2022). Synucleinopathy in Amyotrophic Lateral Sclerosis: A Potential Avenue for Antisense Therapeutics?. International journal of molecular sciences, 23(16), 9364. https://doi.org/10.3390/ijms23169364
- Riancho, J., Gonzalo, I., Ruiz-Soto, M., & Berciano, J. (2019). Why do motor neurons degenerate? Actualization in the pathogenesis of amyotrophic lateral sclerosis. ¿Por qué degeneran las motoneuronas? Actualización en la patogenia de la esclerosis lateral amiotrófica. Neurologia, 34(1), 27–37. https://doi.org/10.1016/j.nrl.2015.12.001
- Saberi, S., Stauffer, J. E., Schulte, D. J., & Ravits, J. (2015). Neuropathology of Amyotrophic Lateral Sclerosis and Its Variants. Neurologic clinics, 33(4), 855–876. https://doi.org/10.1016/j.ncl.2015.07.012
- Pansarasa, O., Bordoni, M., Diamanti, L., Sproviero, D., Gagliardi, S., & Cereda, C. (2018). SOD1 in Amyotrophic Lateral Sclerosis: “Ambivalent” Behavior Connected to the Disease. International journal of molecular sciences, 19(5), 1345. https://doi.org/10.3390/ijms19051345
- Ruffo, P., Perrone, B., & Conforti, F. L. (2022). SOD-1Variants in Amyotrophic Lateral Sclerosis: Systematic Re-Evaluation According to ACMG-AMP Guidelines. Genes, 13(3), 537. https://doi.org/10.3390/genes13030537
- Korobeynikov, V. A., Lyashchenko, A. K., Blanco-Redondo, B., Jafar-Nejad, P., & Shneider, N. A. (2022). Antisense oligonucleotide silencing of FUS expression as a therapeutic approach in amyotrophic lateral sclerosis. Nature medicine, 28(1), 104–116. https://doi.org/10.1038/s41591-021-01615-z
- Roberts, B., Theunissen, F., Mastaglia, F. L., Akkari, P. A., & Flynn, L. L. (2022). Synucleinopathy in Amyotrophic Lateral Sclerosis: A Potential Avenue for Antisense Therapeutics?. International journal of molecular sciences, 23(16), 9364. https://doi.org/10.3390/ijms23169364
- Koch, Y., Helferich, A. M., Steinacker, P., Oeckl, P., Walther, P., Weishaupt, J. H., Danzer, K. M., & Otto, M. (2016). Aggregated α-Synuclein Increases SOD1 Oligomerization in a Mouse Model of Amyotrophic Lateral Sclerosis. The American journal of pathology, 186(8), 2152–2161. https://doi.org/10.1016/j.ajpath.2016.04.008
- Xu, B., Fan, F., Liu, Y., Liu, Y., Zhou, L., & Yu, H. (2023). Distinct Effects of Familial Parkinson’s Disease-Associated Mutations on α-Synuclein Phase Separation and Amyloid Aggregation. Biomolecules, 13(5), 726. https://doi.org/10.3390/biom13050726
- Canever, J. B., Soares, E. S., de Avelar, N. C. P., & Cimarosti, H. I. (2023). Targeting α-synuclein post-translational modifications in Parkinson’s disease. Behavioural brain research, 439, 114204. https://doi.org/10.1016/j.bbr.2022.114204
*Foto de portada creada con la ayuda de DALL·E









