Cáncer de mama. Perspectivas Nacional e Internacional
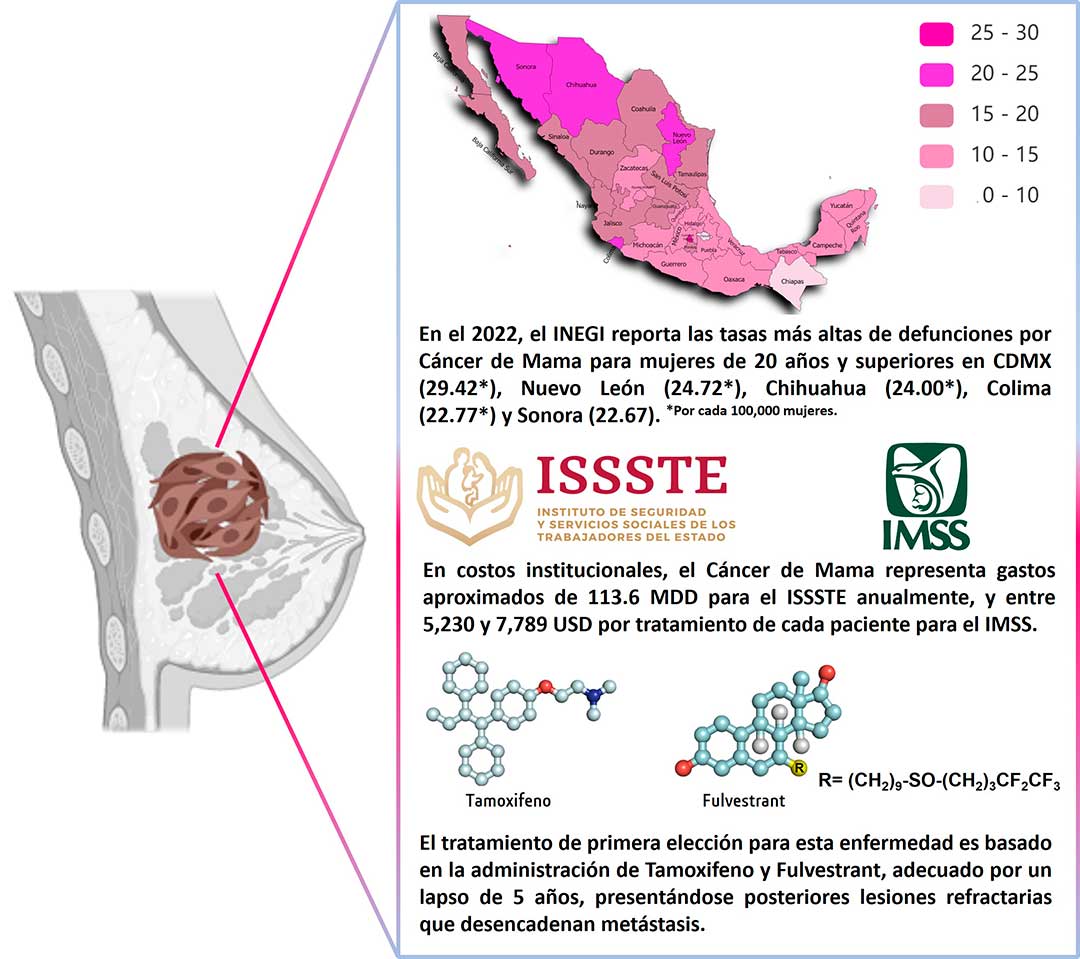
Según la Organización Mundial de la Salud (OMS), el cáncer de mama (CM) fue el más común en 2020, con 2,2 millones de nuevos casos y 685,000 muertes de mujeres en el mundo. En México los números no son alentadores. En el mismo año, el CM fue la enfermedad más frecuente (con 15.3%), siendo la primera causa de muerte en mujeres de entre 30 y 59 años (con 1.52 defunciones por cada 10 mil mujeres).
En costos institucionales, el CM representa inconmensurables gastos a los principales servicios de salud del país. Un estudio del 2017 estimó que el CM representó 3.8% del presupuesto destinado a los servicios de salud prestados por el ISSSTE (Instituto de Seguridad y Servicios Sociales de los Trabajadores del Estado), desglosando en gastos un egreso anual de 113.6 millones de dólares estadounidenses (USD).
En 2023, un estudio realizado en un tercer nivel de atención del IMSS (Instituto Mexicano del Seguro Social), menciona el coste promedio en atención del CM por paciente entre 5,230 y 7,789 USD, para los estadios inicial y avanzado de la enfermedad, reafirmando la importancia del diagnóstico oportuno en aras de evitar un incremento en los gastos del tratamiento en formas avanzadas del padecimiento. (Fig. 1).
Clasificación histológica y molecular del cáncer de mama
La OMS clasifica histológicamente el CM en carcinomas y sarcomas. Los carcinomas pueden ser ductales o lobulares. Ductal cuando el cáncer prolifera en el revestimiento de los conductos mamarios, y lobular cuando crece en los lóbulos de las glándulas mamarias.
Los sarcomas se originan a partir del tejido conectivo, por ejemplo, desde los vasos sanguíneos o los miofibroblastos, soporte estructural de los ductos y los lóbulos. Esta enfermedad también se clasifica empleando marcadores moleculares, cuya expresión depende del componente genético de cada individuo, existiendo cuatro subtipos moleculares: Luminal A, Luminal B, HER2 enriquecido (HER2+) y Triple Negativo (TN).
El grupo de genes responsables de esta diversidad se relacionan con la expresión de receptores de estrógeno (del inglés ERs), de progesterona (del inglés PR), HER2 (receptor del factor de crecimiento epidermal humano 2), y el regulador de la proliferación celular (Ki-67). Lo anterior hace imperante la búsqueda de nuevas opciones terapéuticas basadas en compuestos que interactúen con los blancos farmacológicos de cada uno de los subtipos específicos del CM, disminuyendo los efectos adversos de la radio y quimioterapia contra la enfermedad.
GPER, un nuevo blanco molecular contra el CM
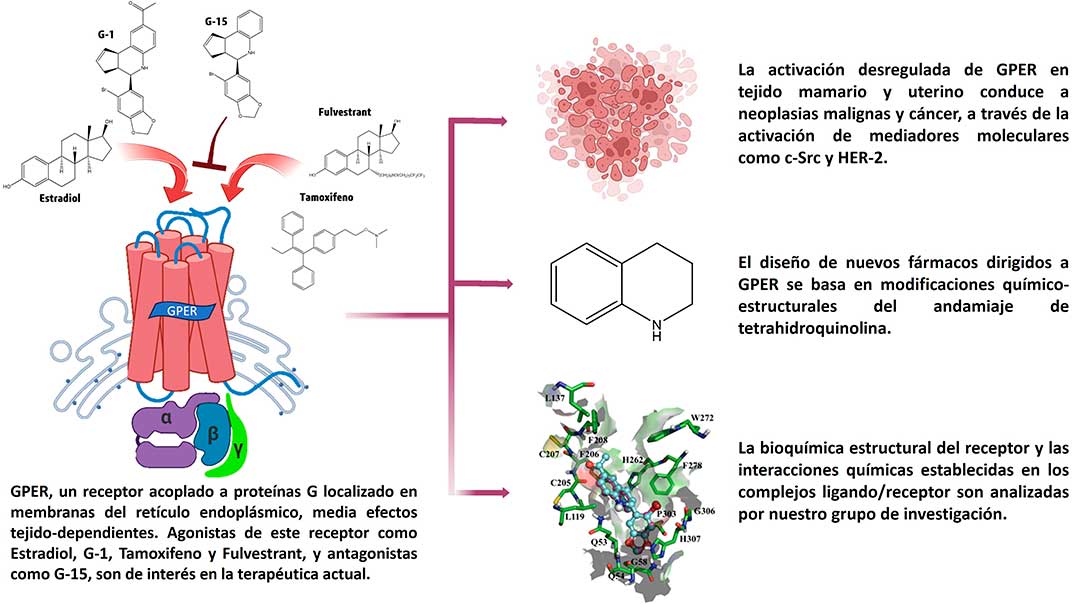
El CM es bioquímicamente complejo, no obstante, con las nuevas tecnologías y la contribución científica, ahora se cuenta con una mejor comprensión sobre su patogénesis molecular, identificando numerosos blancos farmacológicos aptos para la búsqueda de tratamientos más eficaces. Diversos estudios hablan del papel central que desarrolla un receptor transmembranal de estrógenos (GPER) en procesos como la proliferación del CM. GPER, un receptor de la familia de receptores acoplados a proteínas G (del inglés GPCRs), localizado en el retículo endoplásmico y aparato de Golgi, es centro de atención, ya que los fármacos de primera elección para cánceres hormono-sensibles (de respuesta a estrógenos), actúan como agonistas de éste, activando vías de señalización intracelular envueltas en crecimiento y supervivencia del cáncer.
Lo anterior demostró que fármacos como Tamoxifeno y Raloxifeno (moduladores selectivos de los Receptores de Estrógenos, del inglés SERMs), así como Fulvestrant (degradador selectivo del receptor de estrógenos, del inglés SERD), sean agonistas de GPER promoviendo el desarrollo de la enfermedad. Ello abrió la puerta para la investigación encaminada al hallazgo de nuevos fármacos dirigidos al sitio de unión en GPER, como alternativas a las opciones terapéuticas actuales contra el CM (Fig. 2).
Diseño de nuevos fármacos asistido por computadora
Con las nuevas tecnologías, hoy en día, tanto en la academia como en la industria se emplean métodos computacionales para el diseño de fármacos. Éstos se rigen bajo un eje temático de dos estrategias, el diseño de fármacos basado en la estructura (del blanco farmacológico, generalmente una proteína), y el diseño de fármacos basado en ligando (estructura química del ligando afín al receptor). La primera hace uso de información estructural del receptor proveniente de bases de datos como la RCSB-PDB (https://www.rcsb.org/), que cuenta con más de 273, 381 estructuras proteicas resueltas por métodos de difracción de rayos X, resonancia magnética nuclear (RMN), o crio-microscopía electrónica de barrido. Además de información sobre la función de cada componente estructural que conforma la proteína, por ejemplo, longitud en aminoácidos, cofactores, catálisis, dominios de unión a ligando o a otras proteínas, etcétera, encontrándola en el catálogo más completo de secuencias de proteínas y anotaciones funcionales Uniprot (http://www.uniprot.org).
Con el conocimiento estructural del receptor, es posible proponer grupos farmacofóricos (componentes químicos), esencialmente reconocidos por el receptor; diseñar nuevas moléculas (diseño de novo), o reposicionar las ya conocidas para llevar a cabo simulaciones de acoplamiento molecular (del inglés Docking) comparando parámetros energéticos como la energía libre de unión (ΔG en kcal/mol), así como de la naturaleza química de las interacciones establecidas en los complejos ligando/receptor, seleccionando las que mejor se ajusten estructural y energéticamente para ser sintetizados o reposicionados, según corresponda.
La segunda estrategia, se fundamenta en la aplicación de los dos principales métodos para este fin, la técnica de QSAR (del inglés Quatitative Structure Activity Relationship) y el modelado farmacofórico, ambas empleando la información químico-estructural de un grupo de ligandos previamente estudiados desde una perspectiva cuántico-mecánico molecular.
Los datos obtenidos de estas técnicas corresponden a parámetros de reactividad química como la hidrofilicidad, aromaticidad, dureza, blandura, etcétera, para desarrollar métodos de QSAR, o de características farmacofóricas (funcionalidades químicas abstractas en lugar de grupos funcionales específicos), para la construcción de mapas farmacofóricos cuyos requerimientos estéricos sirvan en protocolos de diseño de nuevas moléculas o de reposicionamiento de fármacos.
En la biología del cáncer, se cuenta con las técnicas bioinformáticas necesarias para la implementación y desarrollo de estrategias dirigidas a la búsqueda de nuevos fármacos que, correctamente funcionalizados, se dirijan a algún blanco farmacológico de interés en la terapéutica contra esta enfermedad.
Bioquímica estructural de GPER para el diseño de nuevos fármacos
Empleando métodos computacionales como el modelado y acoplamiento molecular, en estudios previos detectamos el sitio de unión a ligando en GPER, describiendo atómicamente las interacciones químicas establecidas en el complejo ligando/receptor, y los aminoácidos involucrados en el proceso de reconocimiento molecular hacía compuestos con un andamiaje de tetrahidroquinolina como G1 (agonista), G15 y G36 (antagonistas).
Adicionalmente describimos el papel de los aminoácidos Cys207 y Cys130 que, formando un puente disulfuro, se propuso sea el switch molecular del proceso de activación del receptor, según el análisis del modo de unión para ambos ligandos. En dicho proceso se distingue que el sustituyente acetilo de G1 (ausente en G15), establece un puente de hidrógeno con el grupo NH del enlace peptídico de Cys207, lo cual influiría modificando el puente disulfuro que este aminoácido establece con Cys130, en los rearreglos conformacionales de las alfa-hélices 3, 5 y 7 involucrados en la activación del receptor.
Posteriormente, con simulaciones por dinámica molecular, empleando parámetros de simulación lo más cercanos a la realidad bioquímica posible, fueron analizados los complejos ligando/receptor de los estudios previos de acoplamiento molecular, permitiéndonos identificar las modificaciones en el patrón de puentes de hidrógeno para cada uno de los complejos G1/GPER y G15/GPER, así como de la participación de Asn310 en el proceso de estabilización del ligando G15.
Estos resultados ampliaron el conocimiento proteico-estructural con respecto a la caracterización del sitio de unión a ligando en GPER, en un esfuerzo adicional para facilitar el diseño y desarrollo de nuevos fármacos dirigidos a este receptor.
Nuevos ligandos de GPER y su aplicabilidad contra el cáncer
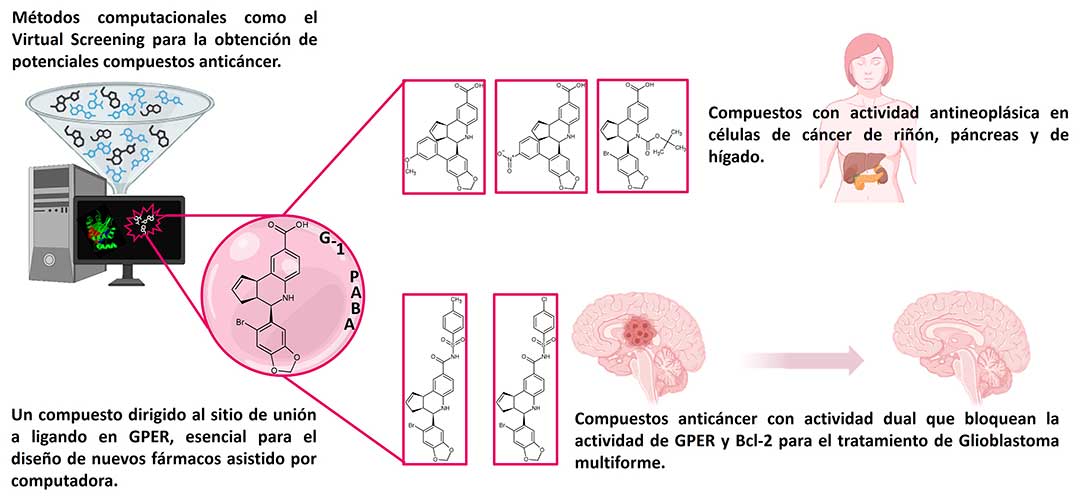
Utilizando el diseño de nuevos fármacos asistido por computadora y partiendo del conocimiento bioquímico-estructural de nuestros estudios previos, obtuvimos 3 compuestos dirigidos al sitio de unión en GPER. La estrategia metodológica para su obtención consistió en la modificación de un análogo del agonista G1 (cuyo grupo acetilo es sustituido desde su síntesis, por un ácido carboxílico) llamado en nuestro grupo de investigación G1-PABA, mediante la sustitución de su átomo de bromo por un anillo aromático para sustituido por un grupo NO2 o un grupo Metoxilo, para los dos primeros compuestos.
El tercer compuesto consistió en la protección del grupo NH del anillo de piperidina empleando el grupo protector Boc (terc-butiloxicarbonilo). (Fig. 2). Estos compuestos se evaluaron en líneas celulares de cáncer, analizando la capacidad de inhibir el crecimiento celular, obteniendo resultados por debajo de los 50 µM en la IC50 (concentración a la que una sustancia ejerce la mitad de su efecto inhibidor máximo), en las líneas RCC4_VA (de carcinoma renal), Mia-Paca-2 (de tumor pancreático) y Hep-G2 (de carcinoma hepatocelular), lo que demostró la importancia fisiopatológica de la expresión de GPER y del bloqueo de su actividad desregulada en otros tipos de cáncer.
En este mismo trabajo, un dato estructural de suma importancia fue la caracterización de los aminoácidos Phe206, Phe278 y Phe314 en el proceso de reconocimiento molecular hacía los compuestos sintetizados, definiéndolos en conjunto como el clúster de fenilalaninas, de importancia fundamental en el diseño de nuevos fármacos dirigidos hacia el sitio de unión en el receptor GPER.
Inhibición dual como un nuevo y prometedor abordaje farmacológico. GPER en glioblastoma
En Glioblastoma multiforme (GBM), un cáncer cerebral de muy mal pronóstico en adultos se ha descrito que GPER desencadena mecanismos de proliferación y supervivencia de células tumorales, lo que torna a este receptor un blanco farmacológico de gran interés para el diseño de nuevos fármacos contra GBM.
Recientemente, aplicando una estrategia dual basada en el bloqueo de dos blancos farmacológicos con el empleo de un mismo fármaco, obtuvimos dos compuestos en cuya estructura química se encuentra el andamiaje de tetrahidroquinolina dirigido a GPER, y porciones químicas del tipo bencen-sulfonamida sustituidas con un átomo de cloro y otro más con un grupo metilo, dirigidos a Bcl-2, una proteína antiapoptótica de significativo interés farmacológico en la investigación contra cáncer. Se identificó un sitio de unión específico, denominado cavidad aceptora de bencen-sulfonamidas, cuya modulación por estos inhibidores selectivos indujeron muerte celular. Los valores de IC50 obtenidos en las líneas celulares de glioblastoma LN-18 y U373 fueron por debajo de 40 µM a 70 µM, respectivamente, representando un resultado alentador como una potencial opción terapéutica para este tipo de neoplasia maligna (Figura 3).
Conclusiones
La investigación de nuevos tratamientos contra el cáncer encuentra en las herramientas computacionales como el modelado, acoplamiento y dinámica molecular, así como el virtual screening, una oportunidad única para proporcionar terapias cada vez más personalizadas, las cuales, dirigidas a un blanco biológico de interés farmacológico, representen alternativas terapéuticas que aseguren una remisión total de la enfermedad, así como un incremento en la expectativa de vida de las personas afectadas.
GPER es un blanco farmacológico cuyo conocimiento creciente sobre la bioquímica estructural de su sitio de unión a ligando, así como de sus posibles mecanismos de activación, nos ha permitido generar compuestos con potencial actividad farmacológica contra cáncer de mama y glioblastoma. Ello pone de manifiesto la importancia crucial del empleo de los métodos computacionales en la generación de nuevas terapias contra enfermedades como el cáncer.
Referencias
Méndez-Luna, D., Bello, M., & Correa-Basurto, J. (2016). Understanding the molecular basis of agonist/antagonist mechanism of GPER1/GPR30 through structural and energetic analyses. The Journal of Steroid Biochemistry and Molecular Biology, 158, 104–116. https://doi.org/10.1016/j.jsbmb.2016.01.001
Méndez-Luna, D., Morelos-Garnica, L. A., García-Vázquez, J. B., Bello, M., Padilla-Martínez, I. I., Fragoso-Vázquez, M. J., González, A. D., De Pedro, N., Gómez-Vidal, J. A., Mendoza-Figueroa, H. L., & Correa-Basurto, J. (2021). Modifications on the Tetrahydroquinoline Scaffold Targeting a Phenylalanine Cluster on GPER as Antiproliferative Compounds against Renal, Liver and Pancreatic Cancer Cells. Pharmaceuticals, 14(1), 49. https://doi.org/10.3390/ph14010049
Morelos-Garnica, L., Guzmán-Velázquez, S., Padilla-Martínez, I., García-Sánchez, J., Bello, M., Bakalara, N., Méndez-Luna, D., & Correa-Basurto, J. (2023). In silico design and cell-based evaluation of two dual anti breast cancer compounds targeting Bcl-2 and GPER. Scientific Reports, 13(1). https://doi.org/10.1038/s41598-023-43860-x
Foto de portada: Thirdman