Los seres vivos tienen estructuras biológicas muy complejas, formadas por diferentes sistemas interconectados entre sí para llevar a cabo sus funciones, mantenerse en equilibrio y en buena salud. Si este balance se llega a romper, se puede pasar a un estado alterado conocido como enfermedad.
Los organismos multicelulares, como el ser humano, cuentan con órganos específicos constituidos por tejidos, y éstos, a su vez, por células, componente básico y fundamental de todo ser vivo, cuyas funciones son realizadas en su mayoría por un tipo particular de biomoléculas, algunas sencillas, otras complejas, dinámicas, versátiles y diversas, llamadas proteínas. Además de éstas, existen otras tres biomoléculas esenciales: carbohidratos, lípidos y ácidos nucleicos.
Si bien estos últimos contienen la información genética de las células y determinan las características primarias de las proteínas, éstas son las responsables de ejercer las funciones de la célula, y suelen ser mucho más diversas en estructura y función. Las proteínas pueden tener diferentes tamaños, formas y tipos. Existen proteínas estructurales, de soporte o contráctiles que le dan forma y motilidad a la célula; otras son de almacenamiento o de defensa, como los anticuerpos; otras pueden ser de transporte como la hemoglobina, y unas más, como las hormonas, actúan como señales entre las células. Las hormonas son como palomas mensajeras que llevan información a lugares distantes del cuerpo. Otras proteínas como las enzimas, son capaces de acelerar y controlar las reacciones químicas que se producen dentro de las células. Las proteínas constituyen más del 50% del peso en seco de un organismo
En 2018 se reportó que tan solo una célula de levadura de cerveza Saccharomyces cerevisiae, contiene 42 millones de moléculas de proteínas y que cada proteína tiene entre 1000 y 10,000 copias; algunas son muy abundantes, con más de medio millón de moléculas, mientras que otras existen en menos de 100 moléculas por célula. Se dice que el número de moléculas de proteínas del cuerpo humano es igual a la cantidad de monedas de un peso que llenarían el Océano Pacífico.
A inicios del siglo XIX los científicos comenzaron a estudiar la composición química de los organismos; Jöns Jacob Berzelius, considerado como el padre de la química sueca, sugirió el término proteína (del griego-proteîos que significa primero o primordial, además de que alude al dios griego Proteo, el cual era capaz de transformarse en cualquier animal), para referirse a una sustancia que al parecer era fundamental para los seres vivos. Después, Gerardus Johannes Mulder, por sugerencia de Berzelius, en 1838 empleó el término proteína por primera vez en su artículo “Sobre la composición de algunas sustancias animales”.
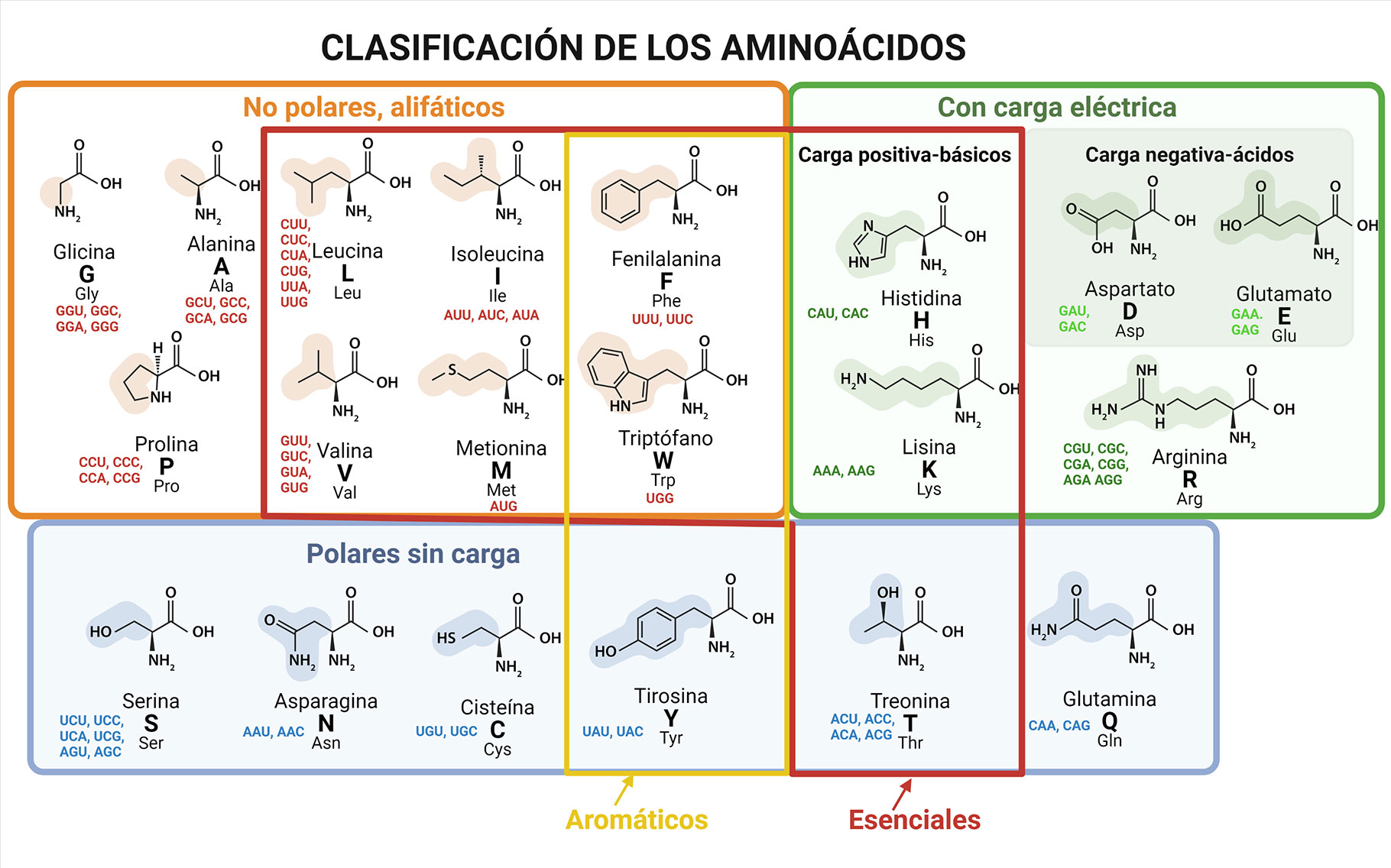
¿De qué están hechas las proteínas? ¿De qué está hecha esta sustancia fundamental? Los aminoácidos son los componentes básicos de las proteínas. En la actualidad, muchos de ellos se utilizan como suplementos alimenticios para aumentar la masa muscular. En 1806, los franceses Louis-Nicolas Vauquelin y Pierre Robiquet, aislaron la asparagina a partir del jugo de espárrago; posteriormente, en 1820, Henri Braconnot, al hervir gelatina descubrió otro aminoácido, la glicina. La lista se completó hacia finales del siglo XIX, cuando se descubrieron y nombraron 20 aminoácidos capaces de formar a las proteínas y cuya composición química fundamental consiste en un átomo de carbono, denominado carbono alfa (α), al cual se le unen un grupo amino (-NH2), un grupo carboxilo (-COOH), un átomo de hidrógeno (-H) y un grupo químico variable al que se le llama cadena lateral o grupo sustituyente (-R), que les confiere propiedades químicas específicas que determinan su identidad y permiten su clasificación en aminoácidos ácidos, básicos, polares y no polares. Es muy importante señalar que 9 de estos 20 aminoácidos deben ingerirse directamente a través de los alimentos, ya que no pueden sintetizarse por las células del ser humano; se designan como aminoácidos esenciales (Figura 1).
Las proteínas tienen diferente secuencia de aminoácidos, que se unen linealmente entre sí mediante un enlace covalente denominado enlace peptídico, donde el grupo carboxilo del aminoácido, reacciona con el grupo amino de otro aminoácido; a esta secuencia se le conoce como estructura primaria y, a pesar de que sólo hay 20 aminoácidos diferentes para formar a las proteínas, la cantidad de combinaciones posibles es prácticamente ilimitada, ya que cada una puede contener varios cientos o miles de aminoácidos.
Para darnos una idea, la titina, una proteína importante en la contracción del músculo estriado, pues permite que las células musculares se contraigan en sintonía, es la proteína más grande conocida del ser humano, compuesta por más de 25,000 aminoácidos cuyo nombre proviene de la palabra Titán (personajes gigantes de la mitología griega con fuerza excepcional).
Esta proteína se sintetiza en el citoplasma usando al ácido ribonucleico (RNA) mensajero (mRNA) que contiene la secuencia de nucleótidos adecuada. Es decir, tiene la información o mensaje para producir esta proteína. El mRNA interactúa con un complejo proteico llamado ribosoma, el cual “lee” la secuencia de nucleótidos del mRNA, usando un triplete de nucleótidos para cada aminoácido, permitiendo que otro tipo de RNA, llamado RNA de transferencia (tRNA), vaya ensamblando los aminoácidos hasta terminar toda la secuencia de mRNA y el ribosoma se detenga cuando encuentre un triplete o codón de paro. Éste no codifica para algún aminoácido y su secuencia puede ser uracilo-guanina-adenina (UGA), uracilo-adenina-guanina (UAG) o uracilo-adenina-adenina (UAA). Una vez terminado el proceso de traducción del mensajero a la secuencia de la proteína (estructura primaria), la proteína naciente debe plegarse y adquirir una conformación tridimensional que la hará funcionalmente activa -proteína activa-, proceso complejo que consta de varios pasos y que involucra a la formación de la estructura secundaria de las proteínas (ESP).
La ESP se refiere a las conformaciones locales de la cadena proteica; las más comunes son las alfa hélices y las láminas beta, que se estabilizan mediante enlaces de hidrógeno. Estas estructuras pueden plegarse o enrollarse de diferente manera y mantenerse unidas por diferentes medios como: las fuerzas de Van der Waals, las interacciones electrostáticas, y los puentes disulfuro (estructura terciaria). Las proteínas también pueden formarse por una o varias cadenas de aminoácidos o polipéptidos (estructura cuaternaria). Sin embargo, estas interacciones pueden interrumpirse por cambios de temperatura, pH, algunos compuestos químicos y otros factores ambientales que pueden alterar su conformación y provocar la destrucción de sus estructuras (proceso conocido como desnaturalización de la proteína) y como consecuencia, causar la pérdida de la función de esa proteína. Un ejemplo claro de este fenómeno es lo que sucede al freír un huevo, donde el calor desnaturaliza a las proteínas ovoalbúminas (proteínas abundantes contenidas en la clara de huevo), que ocasiona la exposición de los aminoácidos hidrofóbicos, los cuales forman una red que la vuelven blanca y opaca.
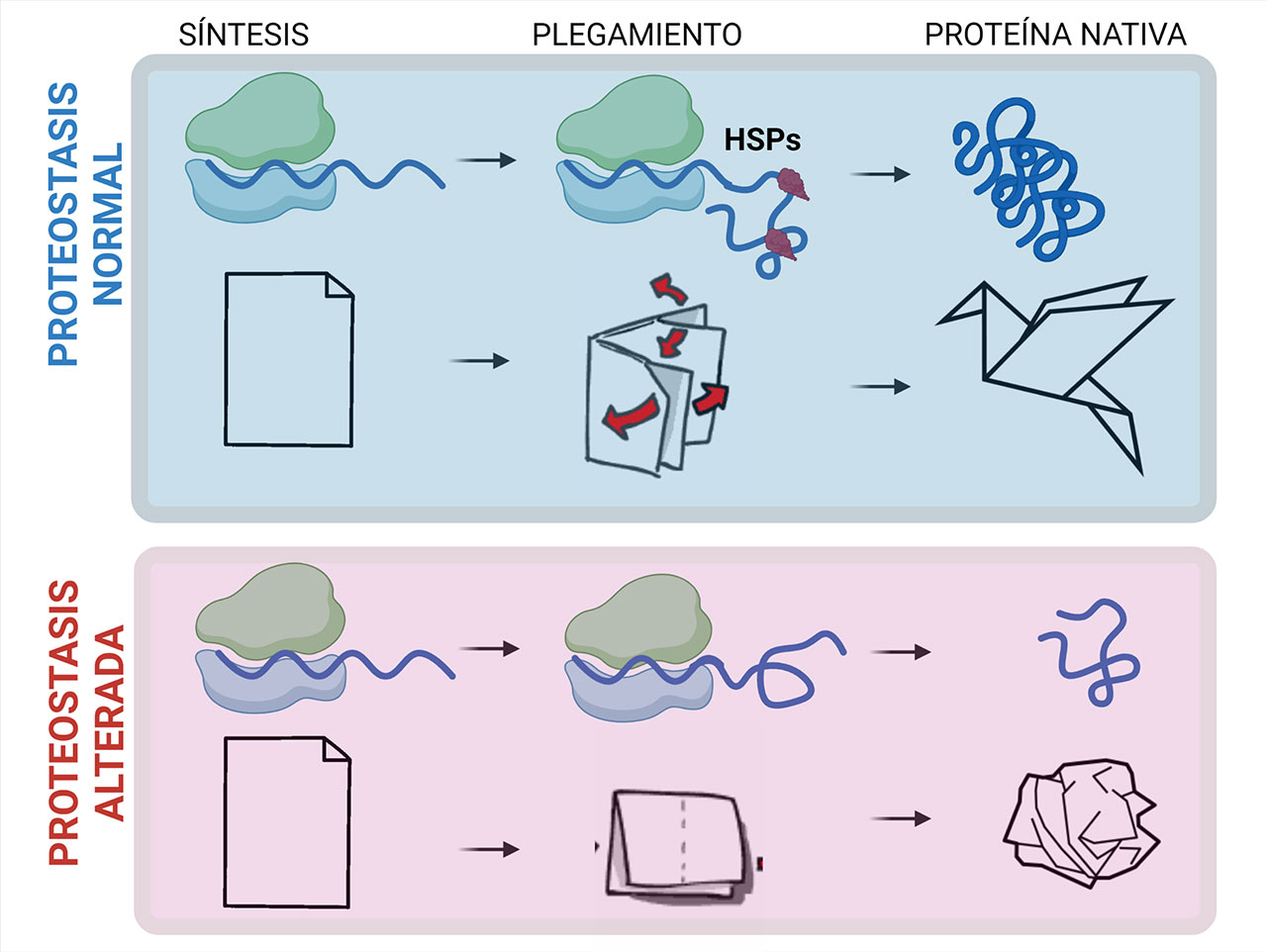
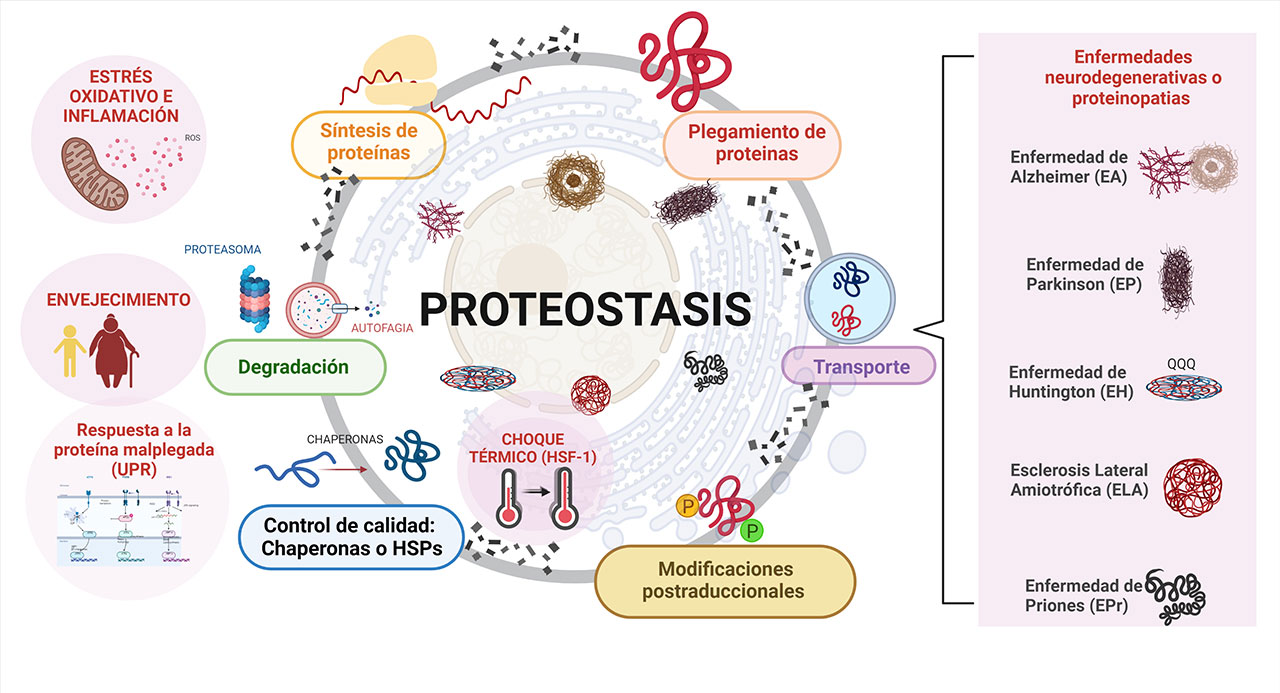
Mantener la estructura funcional de las proteínas no es sencillo. Debido a su procesamiento complejo, deben sujetarse a un mecanismo riguroso de supervisión. La importancia de estas moléculas, su gran diversidad funcional y constante síntesis y recambio, hace que la célula cuente con una gran variedad de medios para controlar que las proteínas se procesen bien y no presenten malos plegamientos o daños que ocasionen alteraciones en su funcionalidad. Al fenómeno por el cual la maquinaria celular mantiene un estricto control de calidad y equilibrio proteico, se le conoce con el nombre de proteostasis, término acuñado por Balch en el 2008 y publicado en la revista Science (1) para referirse a esta compleja red de vías y procesos celulares que mantienen la homeostasis de las proteínas dentro de las células, desde su síntesis, plegamiento, transporte y finalmente su degradación (2). Las proteínas que actúan en varios niveles de esta red de control son las chaperonas, encargadas de llevar a cabo el “origami de las proteínas en las células”. Como escribió David Pincus, las chaperonas moleculares, también conocidas como proteínas de choque térmico (HSPs), son las que facilitan el plegamiento correcto de las proteínas y evitan su agregación, justo como los antiguos chaperones acompañaban a las personas que les encargaban, para vigilar el “buen comportamiento” de las personas a su cuidado.
Además de las chaperonas, existen otros procedimientos de control para eliminar a las proteínas mal plegadas, como son: el sistema ubiquitin-proteasoma y la autofagia; ambos procesos de degradación se encargan de eliminar a las proteínas alteradas. También se tiene a la vía de respuesta a la proteína mal plegada (UPR) que en situaciones de estrés se activa a través de 3 sensores del retículo endoplásmico: la proteína cinasa del retículo endoplásmico (PERK), la cinasa transmembranal endoribonucleasa 1α dependiente de inositol (IRE1α) y el factor de transcripción activador-6 (ATF6), que en condiciones normales están unidos a la proteína de choque térmico BiP manteniéndolas inactivas. Sin embargo, en condiciones de estrés, esta proteína chaperona (BiP) o la proteína de choque térmico (HSPA5), se separan, permitiéndoles activarse por fosforilación inhibiendo la traducción general de proteínas y facilitando la expresión específica de proteínas chaperonas y otros genes protectores de la proteostasis.
Se ha observado que en condiciones de estrés crónico no se puede restablecer la proteostasis, propiciando la agregación de proteínas, lo cual ocasiona toxicidad y muerte celular. Esto es similar a cuando se estira una liga elástica y regresa a su forma original; si se estira demasiado llegará un punto en el que ya no pueda regresar a su forma original, como ocurre con las proteínas de la clara del huevo frito (Figura 2).
Existen varias condiciones patológicas en las que ocurre un desbalance en la proteostasis, se ha reportado que este fenómeno se incrementa durante el envejecimiento y en varios trastornos neurodegenerativos. Esas alteraciones se conocen como proteinopatías y se caracterizan por la acumulación de proteínas mal plegadas en zonas específicas del cerebro, lo que puede provocar sintomatologías diferentes en el individuo afectado. La presencia de estas proteínas desata diferentes procesos fisiopatológicos como son: neuroinflamación, daño oxidativo, activación de las células inmunes del cerebro (microglia), así como alteraciones en los mecanismos de degradación proteínica, y finalmente la muerte neuronal, que puede llevar a la demencia.
La principal demencia conocida hasta ahora es la Enfermedad de Alzheimer (EA) se caracteriza por la acumulación de depósitos extracelulares del péptido amiloide beta (βA) denominados placas amiloides, y por los agregados intracelulares de la proteína tau hiperfosforilada, llamados ovilllos neurofibrilares. Otro padecimiento que presenta grandes acúmulos de proteínas mal plegadas es la enfermedad de Parkinson, la cual se asocia con la acumulación de la proteína alfa-sinucleína en las neuronas dopaminérgicas de la sustancia nigra compacta, llamados cuerpos de Lewy, y están asociados a la muerte de las neuronas dopaminérgicas. De igual manera, en la enfermedad de Huntington, se acumula una forma anormal de la proteína huntingtina. Esta proteína tiene más de 40 glutaminas (polyQ) en su estructura, lo que provoca cambios conformacionales que alteran su función y llegan a causar la muerte neuronal. Otra enfermedad con alteraciones en la acumulación de proteínas es la Esclerosis Lateral Amiotrofica (ELA), en donde la proteína TDP-43 y la superóxido dismutasa 1 (SOD1) se aglomeran en las neuronas motoras y en las células gliales, provocando alteraciones funcionales y la muerte. Finalmente, otro trastorno en donde es muy claro el plegamiento anormal de una proteína es la enfermedad causada por los priones o encefalopatía espongiforme con agregados extracelulares de la proteína priónica (Figura 3).
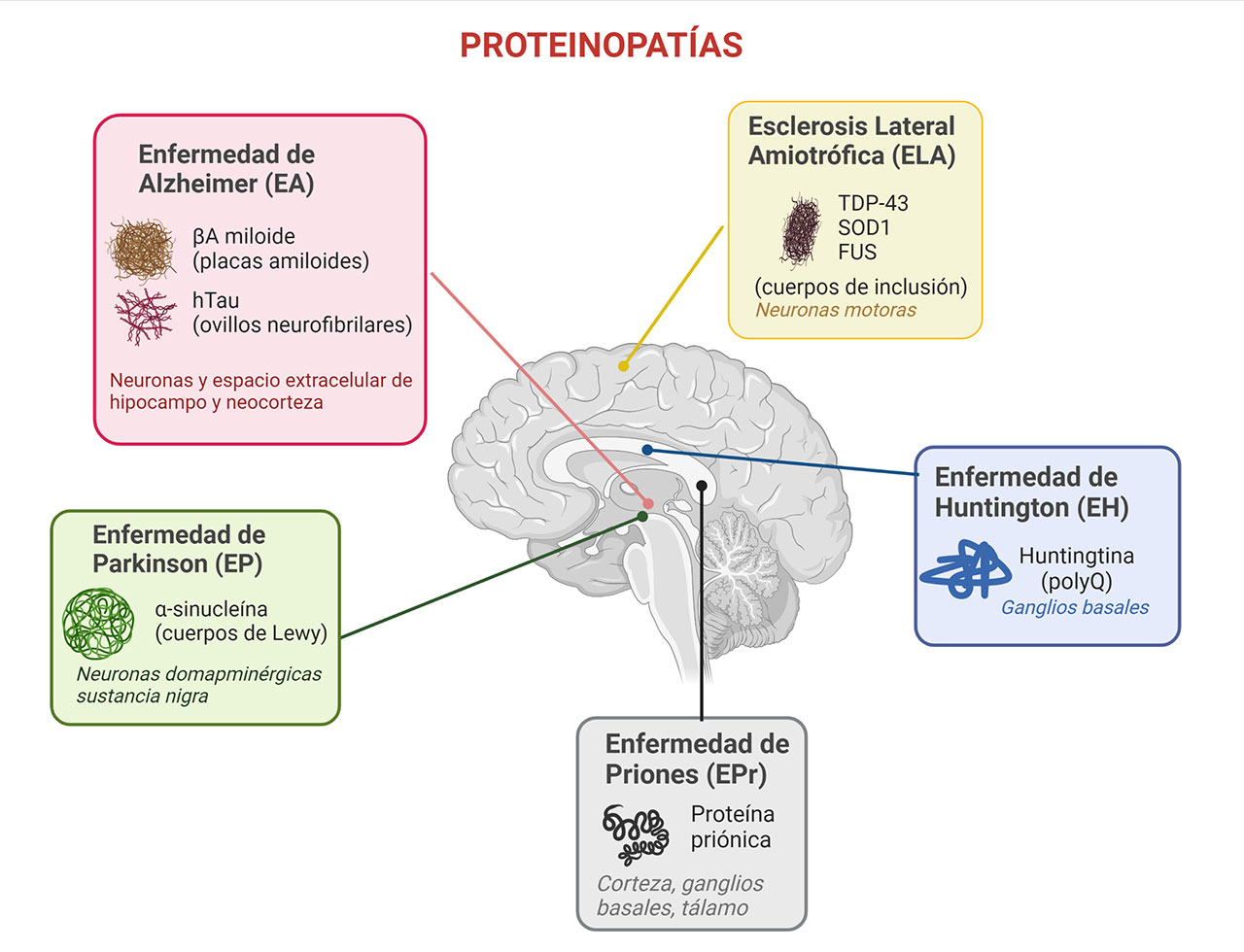
Hasta la fecha no existe un tratamiento eficaz, ni herramientas de diagnóstico adecuadas para la mayoría de estas enfermedades, causadas por proteínas anormales. Sin embargo, diversos estudios demuestran que las células del cerebro de los pacientes son incapaces de formar y degradar adecuadamente a estas proteínas. Se ha observado que una posible causa son los niveles de las proteínas chaperonas —disminuidos o aumentados— y que esto se incrementa con la edad. La modificación en los niveles o composición de las proteínas chaperonas favorece la acumulación de las proteínas alteradas y la formación de estos agregados, generando una alteración en la proteostasis celular. Se ha reportado que cuando se restablecen los niveles normales de las proteínas chaperonas, se puede disminuir e incluso prevenir la agregación de las proteínas alteradas.
El estudio de la proteostasis y los mecanismos que subyacen al procesamiento y plegamiento incorrecto de las proteínas en las distintas enfermedades neurodegenerativas es fundamental. En años recientes, se han reportado avances considerables en este campo y descifrado las estructuras proteicas de muchas proteínas mediante metodologías novedosas como la Cristalografía de Rayos X, la Microscopia Crioelectrónica, la Resonancia Magnética Nuclear (RMN) y se han realizado estudios proteómicos empleando la Espectrometría de Masas. Todas estas tecnologías han permitido identificar, cuantificar y analizar el conjunto completo de proteínas en una célula, en un tejido o en un organismo completo, para evaluar sus funciones, interacciones, modificaciones o cambios específicos y con ello comparar los factores proteínicos (proteomas) de las células sanas y enfermas, y bajo distintas condiciones de estímulos y tratamientos que han permitido desarrollar nuevas estrategias terapéuticas que ayuden a combatir estas enfermedades. Aún queda mucho por descifrar de este origami de proteínas para comprender los mecanismos patológicos que facilitan el desarrollo de las afecciones y con ello, descubrir los fármacos para el tratamiento de estos pacientes o identificar las etapas tempranas de la producción de las proteínas anormales para el diagnóstico oportuno estas afectaciones (Figura 4).
Referencias
- Balch WE, Morimoto RI, Dillin A, Kelly JW. Adapting proteostasis for disease intervention. Science 2008 Feb 15;319(5865):916-9. doi: 10.1126/science.1141448. PMID: 18276881.
- Jayaraj GG, Hipp MS, Hartl FU. Functional Modules of the Proteostasis Network. Cold Spring Harb Perspect Biol. 2020 Jan 2;12(1):a033951. doi: 10.1101/cshperspect.a033951. PMID: 30833457; PMCID: PMC6942124.