La Enfermedad de Alzheimer Familiar (EAF) se presenta en individuos menores a 65 años portadores de mutaciones en los genes de la Proteína Precursora del Amiloide (APP), Presenilina 1 (PSEN1) y Presenilina 2 (PSEN2). La forma esporádica de la Enfermedad de Alzheimer (EA) es multifactorial y se relaciona con la presencia del alelo ε4 de la Apolipoproteína E (APOE ε4), el cual puede incrementar hasta 12 veces el riesgo de desarrollar la enfermedad.

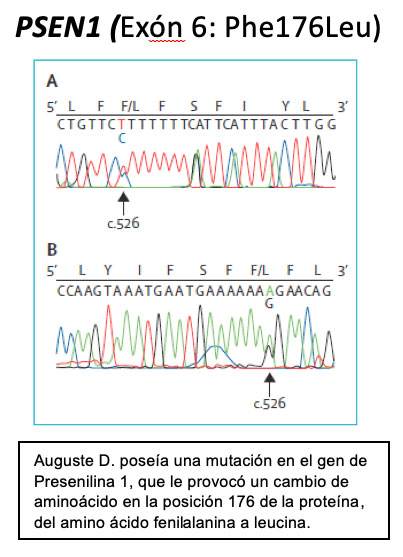
Según las notas de Alois Alzheimer: Cuando Augusta D. llegó al Hospital de Frankfurt en 1902, tenía 51 años y presentaba un cuadro de confusión, ansiedad y agitación. Examinarla, escribía el Dr. Alois, resultaba muy complicado. Una vez que se calmó pudimos realizarle el pertinente examen y concluir que su memoria estaba dañada, tenía dificultades para hablar, se desorientaba y sufría paranoia y alucinaciones auditivas. No supo decir su apellido y por eso decidimos apellidarla con la letra D.
Las mutaciones en el gen de Presenilina 1 (PSEN1) son las más comunes en los casos del Alzheimer Familiar y han sido reportadas al menos 253 mutaciones en este gen (Alzforum, 2019). Estas mutaciones provocan una marcada heterogeneidad del cuadro clínico del individuo portador que, además de tener o no la sintomatología “típica” de la EA, puede cursar con otros padecimientos como parkinsonismo, mioclonía, ataques epilépticos, cambios conductuales parecidos a demencia frontotemporal, afasia y ataxia cerebelosa, así como paraparesia espástica (Larner, 2013; Alzforum, 2019).
En México, se conoce poco sobre cuáles y cuántas mutaciones son responsables de la EAF en los individuos afectados. Recientemente, la mutación que genera un cambio en la posición 431 de la proteína del aminoácido alanina por un ácido glutámico (A431E), ha sido identificada en un gran número de individuos diagnosticados con demencia temprana, la mayoría de ellos originarios del estado de Jalisco, de donde se sugiere que es una mutación con efecto fundador y se ha rastreado en otras familias que radican en el extranjero e incluso, en otras entidades del país, incluida la CDMX (Murrel et al., 2006; Santos-Mandujano et al., 2019).
En 2001, Rogaeva y colaboradores identificaron a la mutación A431E en el gen de PSEN1, junto con otras mutaciones de nuevo registro; fue una de las mutaciones más frecuentes en su grupo de estudio que involucraba a sujetos con una edad promedio de 58 ± 11 años y con un diagnóstico clínico de la EA en su mayoría; sin embargo, no informaron los detalles clínicos ni la historia familiar de los individuos portadores de esta mutación.
 Años más tarde, Yescas et al. (2006) describieron esta mutación en nueve familias mexicanas, aparentemente no relacionadas, con EA de inicio temprano y sugirieron que la mutación se originó en un ancestro común. También encontraron que los análisis de las relaciones genealógicas (o pedigrí), mostraban una transmisión autosómica dominante en al menos tres generaciones, con una edad promedio de presentación de síntomas de 40 años y tenían atrofia subcortical. En este mismo año (Murrel et al. (2006), se identificaron a 20 pacientes adicionales que también portaban la mutación A431E provenientes de 15 familias, 14 de las cuales eran de ascendencia mestiza mexicana y en 9 de ellas, podría rastrearse su ascendencia en el estado de Jalisco y presentaban un cuadro clínico similar a las reportadas por Yescas et al. (2006); algo interesante fue que 9 de los 20 pacientes tenían paraparesia y problemas cognitivos caracterizados por la pérdida de la memoria y tal como lo había reportado previamente, Ringman et al., (2004) se presentaban cambios psicológicos, especialmente de depresión.
Años más tarde, Yescas et al. (2006) describieron esta mutación en nueve familias mexicanas, aparentemente no relacionadas, con EA de inicio temprano y sugirieron que la mutación se originó en un ancestro común. También encontraron que los análisis de las relaciones genealógicas (o pedigrí), mostraban una transmisión autosómica dominante en al menos tres generaciones, con una edad promedio de presentación de síntomas de 40 años y tenían atrofia subcortical. En este mismo año (Murrel et al. (2006), se identificaron a 20 pacientes adicionales que también portaban la mutación A431E provenientes de 15 familias, 14 de las cuales eran de ascendencia mestiza mexicana y en 9 de ellas, podría rastrearse su ascendencia en el estado de Jalisco y presentaban un cuadro clínico similar a las reportadas por Yescas et al. (2006); algo interesante fue que 9 de los 20 pacientes tenían paraparesia y problemas cognitivos caracterizados por la pérdida de la memoria y tal como lo había reportado previamente, Ringman et al., (2004) se presentaban cambios psicológicos, especialmente de depresión.
Un marcador importante de la EA, es la presencia del péptido Amiloide beta (bA) en plasma o líquido céfalo raquídeo (LCR). Al evaluar los niveles de 42 o 40 aminoácidos (βA42 y βA40) en plasma o líquido cefalorraquídeo en estos individuos, no se encontraron diferencias significativas al compararse con sujetos control (Ringman et al. 2008). Sin embargo, la proporción βA42/βA40 fue mayor en las personas portadoras de la mutación. Por otro lado, la otra proteína importante en la EA, la proteína Tau, mostró niveles elevados, así como su estado hiper-fosforilado, en los individuos pre-sintomáticos.
Los estudios de Soosman et al. (2017), usando técnicas de imagenología, mostraron disminución de extensas áreas de materia blanca, subyacente a la corteza motora, en hombres que padecían paraparesis espástica asociada a la mutación en PSEN1 A431E, en comparación con mujeres que poseían otras mutaciones en PSEN1 (incluida A431E) y no presentaban paraparesis espástica. Por otro lado, aunque no observaron diferencias significativas en los volúmenes de las estructuras cerebrales, encontraron que en todos los individuos con paraparesis espástica podían identificarse micro-hemorragias. No hallaron diferencias en los depósitos del péptido βA en la corteza sensomotora y los volúmenes de materia gris tampoco sufrían alteraciones. Esto sugiere que la disminución de la materia blanca en la corteza motora podría explicar las anomalías motoras observadas en estos individuos, y que no dependía, o al menos no únicamente, del metabolismo exclusivo de APP, sino de otros sustratos.
Hasta ahora, el grupo del Dr. Figuera (Dumois-Petersen et al., 2018), es el grupo con mayor contribución en la identificación de individuos portadores de la mutación A431E, puesto que han registrado esta mutación en aproximadamente el 75% (29/39) de los casos diagnosticados con Demencia Familiar de Inicio Temprano en el Centro Médico Nacional de Occidente-IMSS. La edad promedio del inicio de los síntomas es de 43.3 años, con una evolución de la enfermedad de 7.5 años y al morir de 48.9 años. A través de estudios de genealogía, han detectado un total de 147 individuos afectados y también a 304 familiares menores de 48 años considerados en riesgo y a 233 descendientes de estos pacientes en riesgo potencial. Aunque no especifican el cuadro clínico con el que cursan los portadores de la mutación, reportan que ésta provoca una variabilidad en su presentación e invita a la caracterización clínica y molecular de estos pacientes, así como a la falta de un diagnóstico pre-sintomático para los adultos en riesgo, dadas las implicaciones en los tiempos de diagnóstico.
Es de destacar que sólo habían sido reportados casos heterocigotos para esta mutación; sin embargo, el estado homocigoto de la mutación A431E parece conferir un fenotipo de anticipación, con un inicio clínico más temprano y una progresión más rápida de la neurodegeneración (Parker et al., 2019).
Nuestro grupo de investigación ha reportado la caracterización clínica más completa de los individuos portadores de la mutación A431E y propuesto un posible mecanismo molecular del desarrollo de la enfermedad (Santos-Mandujano et al., 2019). Los sujetos afectados incorporan la paraparesis espástica en su cuadro clínico y coinciden con otros estudios donde se presenta una alta desmielinización (Soosman et al. 2017). Hay atrofia cortical grave e hiperintensidades extensas de la materia blanca periventricular.
Dado que los individuos que desarrollan la EAF debida a la mutación A431E son heterocigotos, es decir, un alelo es normal y el otro es mutado (un cromosoma es normal y el otro está mutado), y ambos alelos (genes) se expresan en las células, se podría esperar que la enfermedad se presente en el individuo conforme avanza la edad, si con ese paso del tiempo cambia la expresión de los alelos y llega a dominar la expresión del alelo mutante. Por lo tanto, la cantidad relativa de la proteína PSEN1 mutante y la normal, que se incorpora en el complejo γ-secretasa (complejo donde es funcional la Presenilina 1), así como el potencial de la proteína mutante para afectar a este complejo enzimático, son factores importantes para la comprensión de la EAF causada por la mutación A431E. Por lo anterior, Collins y cols. (2012) generaron una línea celular con la mutación A431E en el gen de PSEN1 en células de neuroglioma humano (H4). Lo que encontraron fue que todas las células que poseían la mutación, sobre-expresan βA42, y así corroboraban el papel que juega esta mutación en el desarrollo de la enfermedad.
En el Departamento de Biomedicina Molecular del Cinvestav-IPN, ofrecemos el servicio de diagnóstico molecular para la Enfermedad de Alzheimer.
Referencias
- Alzforum, Forest Biometrics Research Institute. (2018). MUTATIONS PSEN-1. www.alzforum.org
- Collins C., Katz E., Lee G., Alford V., Williams H., Guillen D., Wu X., Flanagan J., Sjoberg E., Gandy S., Lockhart D. J., Wustman B., Dungan L.B. (2012). Cell-based model for presenilin 1 early-onset Familial Alzheimer’s Disease (EOFAD): dominant negative effects and relative stabilities of Presenilin 1 with EOFAD mutations. Alzheimer’s & Dementia: The Journal of the Alzheimer’s Association 8(4); P241.
- Dumois-Petersen S, Gallegos MP, Magaña T1, Perea FJ, Figuera LE. Frecuencia de la mutación C.1292 C>A en el gene PSEN1, en pacientes con demencia familiar de inicio temprano del Estado de Jalisco. Contacto: luisfiguera@yahoo.com. XXVII Foro Nacional de Investigación en Salud. Investigación en Inflamación la Medicina del Siglo XXI. Congreso del 4 al 7 de septiembre de 2018. Zacatecas, México.
- Larner AJ1. (2013). Presenilin-1 mutations in Alzheimer’s disease: an update on genotype-phenotype relationships. J Alzheimers Dis. 37(4):653-9. doi: 10.3233/JAD-130746.
- Murrell J, Ghetti B, Cochran E, Macias-Islas MA, Medina L, Varpetian A, Cummings JL, Mendez MF, Kawas C, Chui H, Ringman JM. (2006). The A431E mutation in PSEN1 causing familial Alzheimer’s disease originating in Jalisco State, Mexico: an additional fifteen families. Neurogenetics. Nov;7(4):277-9.
- Parker J., Mozaffar T., Messmore A., Deignan J. L., Kimonis V. E. &. Ringman J. M. (2019). Homozygosity for the A431E mutation in PSEN1 presenting with a relatively aggressive phenotype. Neuroscience Letters, 699: 195 – 198 pp.
- Ringman J. M., Diaz-Olavarrieta C., Rodriguez Y., Chavez M., Paz F., Murrell J., Macias M- A., Hill M.& Kawas C. (2004). Female preclinical presenilin-1 mutation carriers unaware of their genetic status have higher levels of depression than their non-mutation carrying kin. Journal of Neurology, Neurosurgery and Psychiatry, 75(3):500 – 502 pp.
- Ringman J. M., Younkin S.G., Pratico D., Seltzer W., Cole G.M., Geschwind D.H., Rodriguez-Agudelo Y., Schaffer B., Fein J., Sokolow S., Rosario E.R., Gylys K.H., Varpetian A., Medina L.D. & Cummings J. L. (2008). Biochemical markers in persons with preclinical familial Alzheimer disease. Neurology, 71 (2): 85 – 95 pp.
- Rogaeva EA, Fafel KC, Song YQ, Medeiros H, Sato C, Liang Y, Richard E, Rogaev EI, Frommelt P, Sadovnick AD, Meschino W, Rockwood K, Boss MA, Mayeux R, St George-Hyslop P. (2001). Screening for PS1 mutations in a referral-based series of AD cases: 21 novel mutations. Neurology. Aug 28;57(4):621-5.
- Santos Mandujano, Rosalía Alejandrina and Marco Antonio Meraz Ríos. (2019). Unvarying Presentation of Atypical Dementia and Spastic Paraparesis in a Mexican Family Carrying the PSEN1 Ala431Glu “Jalisco” Mutation. (Enviado a publicación).
- Soosman S. K., Joseph-Mathurin N., Braskie M. N., Bordelon Y. M., Wharton D., M. Casado, G. Coppola, H. McCallum, M. Nuwer, P. Coutin-Churchman, L. G. Apostolova, T. Benzinger & J. M. Ringman. (2017). Widespread white matter and conduction defects in PSEN1-related spastic paraparesis. Neurobiology of Aging, 47: 201 – 209 pp.
- Yescas, P., Huertas-Vazquez A., Villarreal-Molina M. T., Rasmussen A., Tusié-Luna M. T., López M., Canizales-Quinteros S. y Alonso M. E. (2006). Founder effect for the Ala431Glu mutation of the presenilin 1 gene causing early-onset Alzheimer’s disease in Mexican families. Neurogenetics, 7: 195 – 200 pp.
Mayte Lizeth Padilla Cristerna, Lory Jhenifer Rochin Hernández, Rosalía Alejandrina Santos Mandujano y Marco Antonio Meraz Ríos
Departamento de Biomedicina Molecular, Centro de Investigación y de Estudios Avanzados del IPN (CINVESTAV), CDMX, México.